

We could not find any results for:
Make sure your spelling is correct or try broadening your search.
| Share Name | Share Symbol | Market | Type |
|---|---|---|---|
| Protalix BioTherapeutics Inc | AMEX:PLX | AMEX | Common Stock |
| Price Change | % Change | Share Price | High Price | Low Price | Open Price | Shares Traded | Last Trade | |
|---|---|---|---|---|---|---|---|---|
| 0.00 | 0.00% | 1.71 | 0 | 09:04:43 |
Or
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM
(Mark One)
For the quarterly period ended
OR
For the transition period from to
(Commission file number)
(Exact name of registrant as specified in its charter)
__ | |
(State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) |
|
|
(Address of principal executive offices) | (Zip Code) |
(
(Registrant’s telephone number, including area code)
N/A
(Former name, former address and former fiscal year, if changed since last report)
Securities registered pursuant to Section 12(b) of the Act:
Title of each class | Trading Symbol(s) | Name of each exchange on which registered | ||
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files).
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act:
Large accelerated filer | ☐ | Accelerated filer | ☐ |
⌧ | Smaller reporting company | ||
Emerging growth company |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ◻
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes
On November 1, 2023, approximately
FORM 10-Q
TABLE OF CONTENTS
PART I – FINANCIAL INFORMATION
Item 1. Financial Statements
PROTALIX BIOTHERAPEUTICS, INC.
CONDENSED CONSOLIDATED BALANCE SHEETS
(U.S. dollars in thousands)
(Unaudited)
| September 30, 2023 |
| December 31, 2022 | |||
ASSETS | ||||||
CURRENT ASSETS: | ||||||
Cash and cash equivalents | $ | | $ | | ||
Short-term bank deposits | | | ||||
Accounts receivable – Trade |
| |
| | ||
Other assets |
| |
| | ||
Inventories |
| |
| | ||
Total current assets | $ | | $ | | ||
NON-CURRENT ASSETS: | ||||||
Funds in respect of employee rights upon retirement | $ | | $ | | ||
Property and equipment, net |
| |
| | ||
Deferred income tax asset | | — | ||||
Operating lease right of use assets |
| |
| | ||
Total assets | $ | | $ | | ||
LIABILITIES AND STOCKHOLDERS' EQUITY (NET OF CAPITAL DEFICIENCY) |
|
|
| |||
CURRENT LIABILITIES: |
|
|
| |||
Accounts payable and accruals: |
|
|
| |||
Trade | $ | | $ | | ||
Other |
| |
| | ||
Operating lease liabilities |
| |
| | ||
Contracts liability |
| — |
| | ||
Convertible notes | | — | ||||
Total current liabilities | $ | | $ | | ||
LONG TERM LIABILITIES: |
|
|
| |||
Convertible notes | $ | | ||||
Liability for employee rights upon retirement | $ | |
| | ||
Operating lease liabilities |
| |
| | ||
Total long term liabilities | $ | | $ | | ||
Total liabilities | $ | | $ | | ||
COMMITMENTS | ||||||
STOCKHOLDERS' EQUITY (CAPITAL DEFICIENCY) | | ( | ||||
Total liabilities and stockholders' equity (net of capital deficiency) | $ | | $ | | ||
The accompanying notes are an integral part of the condensed consolidated financial statements.
2
PROTALIX BIOTHERAPEUTICS, INC.
CONDENSED CONSOLIDATED STATEMENTS OF OPERATIONS
(U.S. dollars in thousands, except share and per share data)
(Unaudited)
Nine Months Ended | Three Months Ended | |||||||||||||
| September 30, 2023 |
| September 30, 2022 |
| September 30, 2023 |
| September 30, 2022 | |||||||
REVENUES FROM SELLING GOODS | $ | | $ | | $ | | $ | | ||||||
REVENUES FROM LICENSE AND R&D SERVICES |
| |
| |
| |
| | ||||||
TOTAL REVENUE | | | | | ||||||||||
COST OF GOODS SOLD (1) |
| ( |
| ( |
| ( |
| ( | ||||||
RESEARCH AND DEVELOPMENT EXPENSES (2) |
| ( |
| ( |
| ( |
| ( | ||||||
SELLING, GENERAL AND ADMINISTRATIVE EXPENSES (3) |
| ( |
| ( |
| ( |
| ( | ||||||
OPERATING INCOME (LOSS) |
| |
| ( |
| ( |
| ( | ||||||
FINANCIAL EXPENSES |
| ( |
| ( |
| ( |
| ( | ||||||
FINANCIAL INCOME |
| |
| |
| |
| | ||||||
FINANCIAL INCOME (EXPENSES), NET |
| ( |
| ( |
| |
| ( | ||||||
INCOME (LOSS) BEFORE TAXES ON INCOME | | ( | ( | ( | ||||||||||
TAXES ON INCOME | ( | - | ( | - | ||||||||||
NET INCOME (LOSS) FOR THE PERIOD | $ | | $ | ( | $ | ( | $ | ( | ||||||
EARNINGS (LOSS) PER SHARE OF COMMON STOCK: | ||||||||||||||
BASIC | $ | | $ | ( | $ | ( | $ | ( | ||||||
DILUTED | $ | | $ | ( | $ | ( | $ | ( | ||||||
WEIGHTED AVERAGE NUMBER OF SHARES OF COMMON STOCK | ||||||||||||||
USED IN COMPUTING EARNINGS (LOSS) PER SHARE: | ||||||||||||||
BASIC |
| |
| |
| |
| | ||||||
DILUTED |
| |
| |
| |
| | ||||||
$ | | $ | | $ | | $ | | |||||||
$ | | $ | | $ | | $ | | |||||||
$ | | $ | | $ | | $ | | |||||||
The accompanying notes are an integral part of the condensed consolidated financial statements.
3
PROTALIX BIOTHERAPEUTICS, INC.
CONDENSED CONSOLIDATED STATEMENTS OF CHANGES IN
STOCKHOLDERS’ EQUITY (CAPITAL DEFICIENCY)
(U.S. dollars in thousands, except share data)
(Unaudited)
|
|
| Additional |
|
|
| ||||||||
Common | Common | Paid-In | Accumulated | |||||||||||
Stock (1) | Stock | Capital | Deficit | Total | ||||||||||
Number of | ||||||||||||||
| Shares | Amount | ||||||||||||
Balance at January 1, 2022 |
| | $ | | $ | | $ | ( | $ | ( | ||||
Changes during the nine-month period ended September 30, 2022: |
|
|
|
|
|
|
|
|
| |||||
Issuance of common stock under the Sales Agreement, net |
| | | | | |||||||||
Share-based compensation related to stock options | | | ||||||||||||
Share-based compensation related to restricted stock awards | | * | | | ||||||||||
Exercise of warrants | | * | | | ||||||||||
Net loss for the period |
|
|
|
| ( |
| ( | |||||||
Balance at September 30, 2022 |
| | $ | | $ | | $ | ( | $ | ( | ||||
Balance at January 1, 2023 |
| | $ | | $ | | $ | ( | $ | ( | ||||
Changes during the nine-month period ended September 30, 2023: |
|
|
|
|
|
|
|
|
|
| ||||
Issuance of common stock under the Sales Agreement, net |
| | | | | |||||||||
Convertible notes conversions | | | | | ||||||||||
Share-based compensation related to stock options |
|
|
| |
|
| | |||||||
Share-based compensation related to restricted stock awards |
| | | | ||||||||||
Exercise of warrants |
| | * | | | |||||||||
Net income for the period |
|
|
| | | |||||||||
Balance at September 30, 2023 |
| | $ | | $ | | $ | ( | $ | | ||||
Balance at June 30, 2022 | | $ | | $ | | $ | ( | $ | ( | |||||
Changes during the three-month period ended September 30, 2022: | ||||||||||||||
Issuance of common stock under the Sales Agreement, net | | | | |||||||||||
Share-based compensation related to stock options | | | ||||||||||||
Share-based compensation related to restricted stock awards | | | ||||||||||||
Net loss for the period | ( | ( | ||||||||||||
Balance at September 30, 2022 | | $ | | $ | | $ | ( | $ | ( | |||||
Balance at June 30, 2023 | | $ | | $ | | $ | ( | $ | | |||||
Changes during the three-month period ended September 30, 2023: | ||||||||||||||
Share-based compensation related to stock options | | | ||||||||||||
Share-based compensation related to restricted stock awards | | | | |||||||||||
Net loss for the period | ( | ( | ||||||||||||
Balance at September 30, 2023 | | $ | | $ | | $ | ( | $ | | |||||
*Represents an amount equal to less than $1.
(1)
The accompanying notes are an integral part of the condensed consolidated financial statements.
4
PROTALIX BIOTHERAPEUTICS, INC.
CONDENSED CONSOLIDATED STATEMENTS OF CASH FLOWS
(U.S. dollars in thousands)
(Unaudited)
Nine Months Ended | |||||||
| September 30, 2023 |
| September 30, 2022 | ||||
CASH FLOWS FROM OPERATING ACTIVITIES: |
|
|
|
| |||
Net income (loss) | $ | | $ | ( | |||
Adjustments required to reconcile net income (loss) to net cash used in operating activities: |
|
| |||||
Share-based compensation |
| |
| | |||
Depreciation |
| |
| | |||
Financial income, net (mainly exchange differences) |
| ( |
| ( | |||
Changes in accrued liability for employee rights upon retirement |
| ( |
| ( | |||
Changes in deferred tax asset | ( | — | |||||
Loss (gain) on amounts funded in respect of employee rights upon retirement |
| ( |
| | |||
Gain on conversions of convertible notes | ( | — | |||||
Amortization of debt issuance costs and debt discount | | | |||||
Changes in operating assets and liabilities: |
|
| |||||
Decrease in contracts liability |
| ( |
| ( | |||
Increase in accounts receivable-trade and other assets |
| ( |
| ( | |||
Changes in operating lease right of use assets, net |
| |
| ( | |||
Decrease (increase) in inventories |
| ( |
| | |||
Increase (decrease) in accounts payable and accruals |
| |
| ( | |||
Net cash used in operating activities | $ | ( | $ | ( | |||
CASH FLOWS FROM INVESTING ACTIVITIES: |
|
| |||||
Investment in bank deposits | $ | ( | $ | ( | |||
Proceeds from sale of short-term deposits | | | |||||
Purchase of property and equipment | ( | ( | |||||
Amounts paid (funded) in respect of employee rights upon retirement, net |
| ( |
| | |||
Net cash used in investing activities | $ | ( | $ | ( | |||
CASH FLOWS FROM FINANCING ACTIVITIES: | |||||||
Proceeds from issuance of common stock under the Sales Agreement, net | $ | | $ | | |||
Exercise of warrants | | | |||||
Net cash provided by financing activities | $ | | $ | | |||
EFFECT OF EXCHANGE RATE CHANGES ON CASH AND CASH EQUIVALENTS | $ | ( | $ | ( | |||
NET INCREASE (DECREASE) IN CASH AND CASH EQUIVALENTS |
| |
| ( | |||
BALANCE OF CASH AND CASH EQUIVALENTS AT BEGINNING OF PERIOD |
| |
| | |||
BALANCE OF CASH AND CASH EQUIVALENTS AT END OF PERIOD | $ | | $ | | |||
The accompanying notes are an integral part of the condensed consolidated financial statements.
5
PROTALIX BIOTHERAPEUTICS, INC.
CONDENSED CONSOLIDATED STATEMENTS OF CASH FLOWS
(U.S. dollars in thousands)
(Unaudited)
(Continued) – 2
Nine Months Ended | |||||||
| September 30, 2023 |
| September 30, 2022 | ||||
SUPPLEMENTARY INFORMATION ON INVESTING AND FINANCING ACTIVITIES NOT INVOLVING CASH FLOWS: | |||||||
Purchase of property and equipment | $ | | $ | | |||
Operating lease right of use assets obtained in exchange for new operating lease liabilities | $ | | $ | | |||
Convertible notes conversions | $ | | $ | — | |||
SUPPLEMENTARY DISCLOSURE ON CASH FLOWS |
|
| |||||
Interest paid | $ | | $ | | |||
Interest received | $ | | $ | | |||
The accompanying notes are an integral part of the condensed consolidated financial statements.
6
PROTALIX BIOTHERAPEUTICS, INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(Unaudited)
NOTE 1 - SIGNIFICANT ACCOUNTING POLICIES
a. | General |
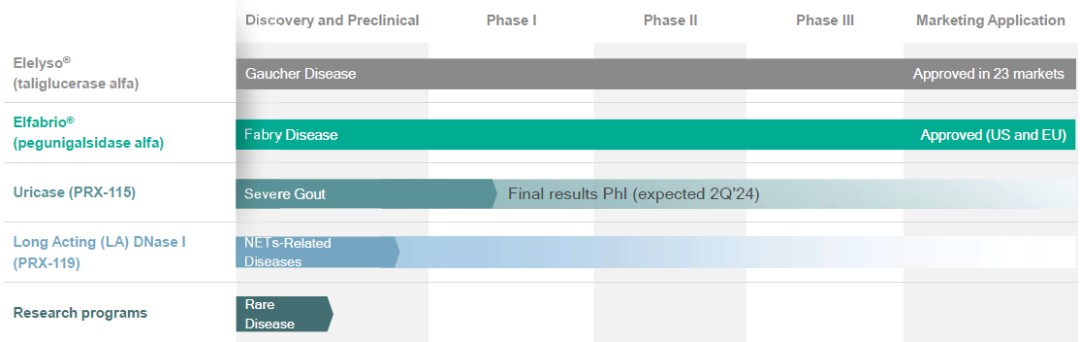
Protalix BioTherapeutics, Inc. (collectively with its subsidiaries, the “Company”) and its wholly-owned subsidiaries, Protalix Ltd. and Protalix B.V. (collectively, the “Subsidiaries”), are biopharmaceutical companies focused on the development and commercialization of recombinant therapeutic proteins based on the Company’s proprietary ProCellEx® protein expression system (“ProCellEx”). The Company’s current focus is to facilitate the commercialization efforts of Chiesi Farmaceutici S.p.A. (“Chiesi”), the Company’s development and commercialization partner for pegunigalsidase alfa, or Elfabrio® (which the Company referred to as PRX-102 during its development stage), for the treatment of Fabry disease, a rare, genetic lysosomal disorder. To date, the Company has successfully developed taliglucerase alfa (marketed under the name Elelyso® except in Brazil where it is marketed as BioManguinhos alfataliglicerase), an enzyme replacement therapy for the treatment of Gaucher disease that has been approved for marketing in the United States, Brazil, Israel and other markets.
The Company’s strategy is to develop proprietary recombinant proteins designed to address high, unmet needs in the rare disease space that are therapeutically superior to existing recombinant proteins currently marketed for the same indications. Consistent with this strategy, the Company has a number of product candidates in varying stages of the clinical development process.
On May 5, 2023, the European Commission (“EC”) announced that it had approved the Marketing Authorization Application (“MAA”) for Elfabrio and on May 9, 2023, the U.S. Food and Drug Administration (“FDA”) announced that it had approved the Biologics License Application (“BLA”) for Elfabrio, each for adult patients with Fabry disease. Both approvals cover the 1 mg/kg every two weeks dosage. The European Medicines Agency (“EMA”) approval followed the February 2023 adoption of a positive opinion and recommendation of marketing authorization for Elfabrio by the EMA’s Committee for Medicinal Products for Human Use the (“CHMP”). Elfabrio was approved by the FDA with a boxed warning for hypersensitivity reactions/anaphylaxis, consistent with Enzyme Replacement Therapy (ERT) class labeling, and Warnings/Precautions providing guidance on the signs and symptoms of hypersensitivity and infusion-associated reactions seen in the clinical studies as well as treatments to manage such events should they occur. The Warnings/Precautions for membranoproliferative glomerulonephritis (MPGN) alert prescribers to the possibility of MPGN and provide guidance for appropriate patient management. Overall, the FDA review team concluded that in the context of Fabry disease as a rare, serious disease with limited therapeutic options that may not be suitable to all individual patients, the benefit-risk of Elfabrio is favorable for the treatment of adults with confirmed Fabry disease.
In August and September of 2023, Elfabrio was approved in Great Britain and Switzerland, respectively, for long-term enzyme replacement therapy in adult patients with a confirmed diagnosis of Fabry disease.
The Company has entered into
Elfabrio, an enzyme replacement therapy, or ERT, was the subject of a phase III clinical program studying the drug as a treatment of patients with Fabry disease, a rare, genetic lysosomal disorder. The phase III clinical program included three separate studies, which are referred to as the BALANCE study, the BRIDGE study and the BRIGHT study. The phase III clinical program analyzed two potential dosing regimens: 1 mg/kg every two weeks and 2 mg/kg every four weeks. In addition, the phase III clinical program included two extension studies in which subjects that participated in our phase I/II clinical trials and phase III clinical trials had the opportunity to enroll and continue to be treated with PRX-102. As of March 1, 2023, sponsorship of the two open-label extension studies was transferred to Chiesi, which is now administering the extension studies. Over time, and as Elfabrio is approved for marketing in different jurisdictions,
7
PROTALIX BIOTHERAPEUTICS, INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(Unaudited)
participants withdraw from the open-label extension studies. Some of the withdrawals transfer to a commercial setting, others withdraw for other reasons.
The BLA for Elfabrio for the treatment of adult patients with Fabry disease was resubmitted to the FDA on November 9, 2022. An initial BLA for Elfabrio was submitted to the FDA on May 27, 2020 under the FDA’s Accelerated Approval pathway, but resulted in a Complete Response Letter (“CRL”).
The MAA was submitted to the EMA on February 7, 2022, after the October 8, 2021 meeting we held, together with Chiesi, with the Rapporteur and Co-Rapporteur of the EMA regarding PRX-102.
The FDA publicly released the internal review documents for Elfabrio (pegunigalsidase alfa-iwxj) injection BLA 761161. These documents provide previously unavailable additional information regarding the basis for the FDA’s May 2023 approval decision. In particular, the FDA determined that substantial evidence of effectiveness for Elfabrio in Fabry patients was established with one adequate and well-controlled study (Study PB-102-F01/02) with confirmatory evidence provided by the BALANCE study (also referred to as Study PB-102-F20). The FDA review team also concluded that the BALANCE study met its primary efficacy endpoint, which assessed the annualized rate of change in eGFR (estimated glomerular filtration rate) over 104 weeks. However, the FDA also determined that the results from the BALANCE study did not support a non-inferiority claim to the comparator product due to the lack of data to support a non-inferiority margin.
The Company has licensed the rights to commercialize taliglucerase alfa worldwide (other than Brazil) to Pfizer Inc. (“Pfizer”), and in Brazil to Fundação Oswaldo Cruz (“Fiocruz”), an arm of the Brazilian Ministry of Health (the “Brazilian MoH”). Otherwise, except with respect to taliglucerase alfa and Elfabrio, the Company holds the worldwide commercialization rights to its other proprietary development candidates. In addition, the Company continuously evaluates potential strategic marketing partnerships as well as collaboration programs with biotechnology and pharmaceutical companies and academic research institutions.
The Company’s product pipeline currently includes, among other candidates:
On March 21, 2023, the first patient was dosed in the Company’s phase I First-in-Human (“FIH”) clinical trial of PRX-115. As of September 30, 2023,
Obtaining marketing approval with respect to any product candidate in any country is dependent on the Company’s ability to implement the necessary regulatory steps required to obtain such approvals, and demonstrate the safety and efficacy of its product candidates. The Company cannot reasonably predict the outcome of these activities.
On July 2, 2021, the Company entered into an At The Market Offering Agreement (the “2021 Sales Agreement”) with H.C. Wainwright & Co., LLC, as the Company’s sales agent (the “Agent”) which was amended on May 2, 2022. Pursuant to the terms of the 2021 Sales Agreement, the Company was able to sell, from time to time through the Agent, shares of its common stock, par value $
During the term of the 2021 Sales Agreement which ended during the quarter ended March 31, 2023, the Company sold a total of
8
PROTALIX BIOTHERAPEUTICS, INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(Unaudited)
On February 27, 2023, the Company entered into an At The Market Offering Agreement (the “2023 Sales Agreement”) with the Agent. Pursuant to the terms of the 2023 Sales Agreement, the Company may sell, from time to time through the Agent, ATM Shares having an aggregate offering price of up to $
Under each of the Chiesi Ex-US Agreement and the Chiesi US Agreement (collectively, the “Chiesi Agreements”), Chiesi made an upfront payment to Protalix Ltd. of $
Under the terms of both of the Chiesi Agreements, Protalix Ltd. is required to manufacture all of the Elfabrio drug substance needed under the agreements, subject to certain exceptions, and Chiesi will purchase Elfabrio drug product from Protalix, subject to certain terms and conditions. The consideration for Protalix Ltd. is based on the drug product supplied to Chiesi and the average selling price of the drug product in the relevant territory multiplied by tiered payments as described in the relevant agreement. Under the Chiesi Ex-US Agreement, Chiesi is required to make tiered payments for drug product purchased from Protalix of
On August 29, 2022, the Company entered into a Fill/Finish Agreement (the “F/F Agreement”) and a Letter Agreement (the “Letter Agreement”), in each case with Chiesi. The Company agreed to supply Chiesi with drug substance for PRX-102 and, following relevant technology and technical information transfer activities, Chiesi has agreed, among other things, to provide the Company with commercial fill/finish services for PRX-102, including to support the anticipated global launch of PRX-102. The F/F Agreement shall continue in force until December 31, 2025, unless terminated earlier in accordance with the terms of the F/F Agreement and the term may be extended by mutual agreement for an additional period of
On May 13, 2021, the Company signed a binding term sheet with Chiesi pursuant to which the Company and Chiesi amended the Chiesi Agreements in order to provide the Company with near-term capital. Chiesi agreed to make a $
Since its approval by the FDA, taliglucerase alfa has been marketed by Pfizer in accordance with the exclusive license and supply agreement entered into between Protalix Ltd. and Pfizer, which is referred to herein as the Pfizer Agreement. In October 2015, Protalix Ltd. and Pfizer entered into an amended exclusive license and supply agreement, which is referred to herein as the Amended Pfizer Agreement, pursuant to which the Company sold to Pfizer its share in the collaboration created under the Pfizer Agreement for the commercialization of Elelyso. As part of the sale, the Company agreed to transfer its rights to Elelyso in Israel to Pfizer while gaining full rights to it in Brazil. Under the Amended Pfizer Agreement, Pfizer is entitled to all of the revenues, and is responsible for
On June 18, 2013, the Company entered into a Supply and Technology Transfer Agreement with Fiocruz (the “Brazil Agreement”) for taliglucerase alfa. Fiocruz’s purchases of BioManguinhos alfataliglicerase to date have been significantly below certain agreed-upon purchase milestones and, accordingly, the Company has the right to terminate the Brazil Agreement. Notwithstanding the termination right, the Company is, at this time, continuing to supply
9
PROTALIX BIOTHERAPEUTICS, INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(Unaudited)
BioManguinhos alfataliglicerase to Fiocruz and patients continue to be treated with BioManguinhos alfataliglicerase in Brazil.
The Company expects to continue to incur significant expenditures in the near future due to research and developments efforts with respect to the product candidates. Under the terms of the Company’s outstanding
b. | Basis of presentation |
The accompanying unaudited condensed consolidated financial statements of the Company have been prepared in accordance with generally accepted accounting principles in the United States (“GAAP”) for interim financial information. Accordingly, they do not include all of the information and notes required by GAAP for annual financial statements. In the opinion of management, all adjustments (of a normal recurring nature) considered necessary for a fair statement of the results for the interim periods presented have been included. Operating results for the interim period are not necessarily indicative of the results that may be expected for the full year.
These unaudited condensed consolidated financial statements should be read in conjunction with the audited consolidated financial statements in the Annual Report on Form 10-K for the year ended December 31, 2022, filed by the Company with the U.S. Securities and Exchange Commission (the “Commission”). The comparative balance sheet at December 31, 2022 has been derived from the audited financial statements at that date. There have been no material changes in our significant accounting policies as described in our consolidated financial statements for the year ended December 31, 2022.
c. | Revenue recognition |
The Company accounts for revenue pursuant to Accounting Standards Codification, Topic 606, Revenue from Contracts with Customers (“ASC 606”). Under ASC 606, a contract with a customer exists only when: the parties to the contract have approved it and are committed to perform their respective obligations, the Company can identify each party’s rights regarding the distinct goods or services to be transferred (“performance obligations”), the Company can determine the transaction price for the goods or services to be transferred, the contract has commercial substance and it is probable that the Company will collect the consideration to which it will be entitled in exchange for the goods or services that will be transferred to the customer.
Revenues are recorded in the amount of consideration to which the Company expects to be entitled in exchange for performance obligations upon transfer of control to the customer.
1. Revenues from selling products
The Company recognizes revenues from selling goods at a point in time when control over the product is transferred to customers (upon delivery), at the net selling price, which reflects reserves for variable consideration, potential discounts and allowances.
The transaction price is the consideration to which the Company expects to be entitled from the customer. The consideration promised in a contract with the Company’s customers may include fixed amounts and variable amounts. The Company estimates the variable consideration and includes it in the transaction price using the most likely outcome method, and only to the extent it is highly probable that a significant reversal of cumulative revenue recognized will not occur when the uncertainty associated with the variable consideration is subsequently resolved. Prior to recognizing revenue from variable consideration, the Company uses significant judgment to determine the probability of significant reversal of such revenue.
10
PROTALIX BIOTHERAPEUTICS, INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(Unaudited)
2. | Revenues from Chiesi Agreements |
The Company has identified
The Company determined that the license together with the research and development services should be combined into single performance obligation since Chiesi cannot benefit from the license without the research and development services. The research and development services are highly specialized and are dependent on the supply of the drug.
The manufacturing was contingent on regulatory approvals of the drug and the Company deems these services to be separately identifiable from other performance obligations in the contract. Manufacturing services post-regulatory approval are not interdependent or interrelated with the license, research and development services. Following the regulatory approvals for Elfabrio received in May 2023, the Company started recognizing revenue from manufacturing, see also revenue from selling products above.
The transaction price was comprised of fixed consideration and variable consideration (capped research and development reimbursements). Under ASC 606, the consideration to which the Company would be entitled upon the achievement of contractual milestones, which are contingent upon the occurrence of future events, are a form of variable consideration. The Company estimates variable consideration using the most likely method. Amounts included in the transaction price are recognized only when it is probable that a significant reversal of cumulative revenues will not occur. Prior to recognizing revenue from variable consideration, the Company uses significant judgment to determine the probability of significant reversal of such revenue. Following the approval of Elfabrio by the FDA, the Company received a milestone payment equal to $
Since the customer benefits from the research and development services as the entity performs the service, revenue from granting the license and the research and development services was recognized over time using the cost-to-cost method.
Revenue from additional research and development services ordered by Chiesi is recognized over time using the cost-to-cost method.
3. | Revenue from R&D services |
Revenue from the research and development services was recognized over time using the cost-to-cost method since the customer benefits from the research and development services as the entity performs the services.
NOTE 2 - INVENTORIES
Inventories at September 30, 2023 and December 31, 2022 consisted of the following:
| September 30, |
| December 31, | ||||
(U.S. dollars in thousands) | 2023 | 2022 | |||||
Raw materials | $ | | $ | | |||
Work in progress |
| | | ||||
Finished goods |
| | | ||||
Total inventory | $ | | $ | | |||
NOTE 3 – FAIR VALUE MEASUREMENT
The Company measures fair value and discloses fair value measurements for financial assets and liabilities. Fair value is based on the price that would be received from the sale of an asset, or paid to transfer a liability, in an orderly transaction between market participants at the measurement date.
11
PROTALIX BIOTHERAPEUTICS, INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(Unaudited)
The accounting standard establishes a fair value hierarchy that prioritizes observable and unobservable inputs used to measure fair value into three broad levels, which are described below:
Level 1: Quoted prices (unadjusted) in active markets that are accessible at the measurement date for assets or liabilities. The fair value hierarchy gives the highest priority to Level 1 inputs.
Level 2: Observable prices that are based on inputs not quoted on active markets, but corroborated by market data.
Level 3: Unobservable inputs are used when little or no market data is available. The fair value hierarchy gives the lowest priority to Level 3 inputs.
In determining fair value, the Company utilizes valuation techniques that maximize the use of observable inputs and minimize the use of unobservable inputs to the extent possible and considers counterparty credit risk in its assessment of fair value.
The fair value of the financial instruments included in the working capital of the Company is usually identical or close to their carrying value.
Based on a Level 3 measurement, as of September 30, 2023, the fair value of the $
| 2024 Notes | |||||
Stock price (USD) |
| | ||||
Expected term |
| | ||||
Risk free rate |
| | % | |||
Volatility |
| | % | |||
Yield |
| | % |
NOTE 4 – REVENUES
The following table summarizes the Company’s disaggregation of revenues:
| Three Months Ended September 30, | Nine Months Ended September 30, | |||||||||||
(U.S. dollars in thousands) |
| 2023 |
| 2022 | 2023 |
| 2022 | ||||||
Pfizer | $ | | $ | | $ | | $ | | |||||
Brazil | $ | | $ | | $ | | $ | | |||||
Chiesi | $ | | $ | | $ | | $ | | |||||
Total revenues from selling goods | $ | | $ | | $ | | $ | | |||||
Revenues from license and R&D services | $ | | $ | | $ | | $ | | |||||
NOTE 5 – STOCK TRANSACTIONS
| (a) | Authorized Capital |
On June 28, 2023, the Company held its 2023 Annual Meeting of Stockholders, which was adjourned and reconvened on July 13, 2023 (the “Annual Meeting”). At the Annual Meeting, the Company’s stockholders, among other matters, approved an amendment to the Company’s Certificate of Incorporation, as amended, to increase the number of shares of Common Stock authorized for issuance from
12
PROTALIX BIOTHERAPEUTICS, INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(Unaudited)
| (b) | At-the-Market (ATM) Offering |
During the nine months ended September 30, 2023, the Company sold, in the aggregate,
(c)Exercise of Warrants
On May 8, 2023, the Company issued
On May 10, 2023, the Company issued
(d)Conversion of 2024 Notes
During the nine months ended September 30, 2023, the Company issued, in the aggregate,
(e)Stock based compensation
| 1) | On June 28, 2023, at the Annual Meeting, the Company’s stockholders, among other matters, adopted amendments to the Company’s Amended and Restated 2006 Employee Stock Incentive Plan, as amended (the “Plan”) to increase the number of shares of Common Stock available under Plan from |
| 2) | On August 15, 2023, the Company granted, with the approval of the Company’s compensation committee, |
3) | On August 15, 2023, the Company granted, with the approval of the Company’s compensation committee, |
13
PROTALIX BIOTHERAPEUTICS, INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(Unaudited)
On September 14, 2023, the Company granted, with the approval of the Company’s compensation committee,
On September 29, 2023, the Company granted, with the approval of the Company’s compensation committee,
The fair value of each option granted during the three-month period ended September 30, 2023 is estimated at the date of grant using the Black-Scholes option-pricing model. The following weighted average assumptions were applied in determining the fair value of the options described in this Note 5(e)(3) on their respective grant dates:
Weighted average grants date fair value (USD) | | ||
Exercise price (USD) | | ||
Risk free rate | | % | |
Volatility | | % | |
Dividend yield | % | ||
Expected life (Years) |
NOTE 6 – EARNINGS (LOSS) PER SHARE
Basic earnings (loss) per share is calculated by dividing the net income (loss) by the weighted average number of shares of Common Stock outstanding during each period.
Diluted earnings per share is calculated by dividing the net income by the weighted-average number of shares of Common Stock outstanding during each period increased to include the number of additional shares of Common Stock that would have been outstanding if the potentially dilutive shares had been issued.
In computing diluted earnings per share, basic earnings per share are adjusted to take into account the potential dilution that could occur upon: (i) the exercise of options granted under employee stock compensation plans using the treasury stock method; (ii) the exercise of warrants using the treasury stock method; and (iii) the conversion of the convertible notes using the “if-converted” method.
14
PROTALIX BIOTHERAPEUTICS, INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(Unaudited)
Basic and diluted net earnings (loss) per share attributable to common stockholders were calculated as follows:
Three Months Ended September 30, | Nine Months Ended September 30, | |||||||||||
(In thousands, except share data) | 2023 | 2022 | 2023 | 2022 | ||||||||
Numerator: | ||||||||||||
Net income (loss) | $ | ( | $ | ( | $ | | $ | ( | ||||
Add: | ||||||||||||
Financial expenses of 2024 Notes* | $ | ( | — | $ | ( | — | ||||||
Net income (loss) for diluted calculation | $ | ( | $ | ( | $ | | $ | ( | ||||
Denominator: | ||||||||||||
Weighted average shares of Common Stock outstanding for basic calculation | | | | | ||||||||
Weighted average dilutive effect of 2024 Notes | | — | | — | ||||||||
Weighted average dilutive effect of stock options | — | — | | — | ||||||||
Weighted average shares of Common Stock outstanding for diluted calculation | | | | | ||||||||
* Financial expenses on 2024 Notes consists of add back of financial expense incurred during the period and inclusion of make-whole interest payments that will be incurred upon conversion.
Diluted earnings (loss) per share do not include
Diluted earnings (loss) per share do not include
NOTE 7 – TAXES ON INCOME
The following table summarizes the Company’s taxes on income:
| Three Months Ended |
| Nine Months Ended | |||
(U.S. dollars in thousands) | September 30, 2023 | September 30, 2023 | ||||
Current taxes on income | $ | | $ | | ||
Deferred taxes on income |
| | ( | |||
Total taxes on income | $ | | $ | | ||
The Company had an effective tax rate of
Following the regulatory approvals for Elfabrio in May 2023, the receipt of the $
15
PROTALIX BIOTHERAPEUTICS, INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(Unaudited)
Section 174 of the TCJA which requires the Company to capitalize and amortize its research and development expenses over 15 years; and (iii) its forecasted profits in the United States following the regulatory approvals of Elfabrio.
NOTE 8 – SUPPLEMENTARY FINANCIAL STATEMENT INFORMATION
Balance sheets:
| September 30, |
| December 31, | |||
(U.S. dollars in thousands) | 2023 | 2022 | ||||
Accounts payable and accruals – other: | ||||||
Payroll and related expenses | $ | | $ | | ||
Interest Payable | | | ||||
Provision for vacation | | | ||||
Accrued expenses | | | ||||
Royalties payable | | | ||||
Income tax payable | | | ||||
Reserve for deductions from revenue |
| | — | |||
Property and equipment suppliers |
| | | |||
$ | | $ | | |||
NOTE 9 – SUBSEQUENT EVENTS
1) | On October 4, 2023 the Company collected approximately $ |
2) | On October 15, 2023, the Company granted, with the approval of the Company’s compensation committee, |
3) | Because the Company's operations are conducted in the State of Israel, the business and operations may be directly affected by economic, political, geopolitical and military conditions in Israel. In October 2023, Hamas terrorists infiltrated Israel’s southern border from the Gaza Strip and conducted a series of attacks on civilian and military targets. Hamas also launched extensive rocket attacks on Israeli population and industrial centers located along Israel’s border with the Gaza Strip and in other areas within the State of Israel attacking a number of civilian and military targets while simultaneously launching extensive rocket attacks on the Israeli population and industrial centers. At the same time, clashes between Israel and Hezbollah in Lebanon have increased. In response, Israel’s security cabinet declared war against the Hamas and a military campaign against these terrorist organizations commenced in parallel to their continued rocket and terror attacks. Moreover, the attacks by Hamas and Hezbollah, and Israel’s defensive measures, may result in a greater regional conflict. It is currently not possible to predict the duration or severity of the ongoing conflict or its effects on our business, operations and financial conditions. The ongoing conflict is rapidly evolving and developing, and could disrupt certain of our business and operations, among others. |
16
Item 2. Management’s Discussion and Analysis of Financial Condition and Results of Operations
CAUTIONARY STATEMENT REGARDING FORWARD-LOOKING STATEMENTS
AND RISK FACTORS SUMMARY
You should read the following discussion and analysis of our financial condition and results of operations together with our financial statements and the consolidated financial statements and the related notes included elsewhere in this Form 10-Q and in our Annual Report on Form 10-K for the year ended December 31, 2022. Some of the information contained in this discussion and analysis, particularly with respect to our plans and strategy for our business and related financing, includes forward-looking statements within the meanings of Section 27A of the Securities Act and Section 21E of the Securities Exchange Act of 1934, as amended, or the Exchange Act, including statements regarding expectations, beliefs, intentions or strategies for the future. When used in this report, the terms “anticipate,” “believe,” “estimate,” “expect,” “can,” “continue,” “could,” “intend,” “may,” “plan,” “potential,” “predict,” “project,” “should,” “will,” “would” and words or phrases of similar import, as they relate to our company or our management, are intended to identify forward-looking statements. We intend that all forward-looking statements be subject to the safe-harbor provisions of the Private Securities Litigation Reform Act of 1995. These forward-looking statements are only predictions and reflect our views as of the date they are made with respect to future events and financial performance, and we undertake no obligation to update or revise, nor do we have a policy of updating or revising, any forward-looking statement to reflect events or circumstances after the date on which the statement is made or to reflect the occurrence of unanticipated events, except as may be required under applicable law. Forward-looking statements are subject to many risks and uncertainties that could cause our actual results to differ materially from any future results expressed or implied by the forward-looking statements as a result of several factors including those set forth under “Risk Factors” in our Quarterly Report on Form 10-Q for the quarter ended June 30, 2023 and in this Quarterly Report on Form 10-Q.
Examples of the risks and uncertainties include, but are not limited to, the following:
17
Given these uncertainties, you should not place undue reliance on these forward-looking statements. Companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in advanced or late-stage clinical trials, even after obtaining promising earlier trial results or preliminary findings for such clinical trials. Even if favorable testing data is generated from clinical trials of a drug product, the FDA or foreign regulatory authorities may not accept or approve a marketing application filed by a pharmaceutical or biotechnology company for the drug product.
Recent Company Developments
18
ProCellEx: Our Proprietary Protein Expression System
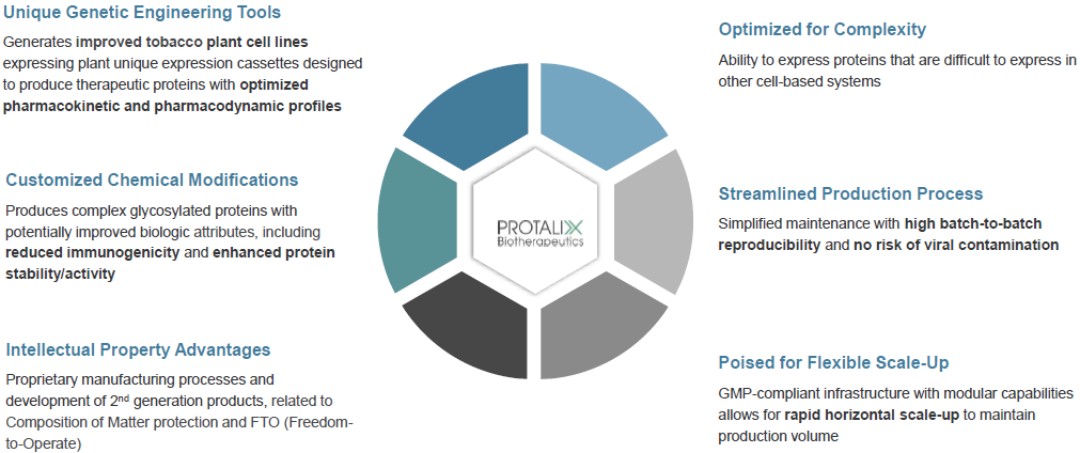
ProCellEx is our proprietary platform used to produce and manufacture recombinant proteins through plant cell-based expressions in suspension. ProCellEx consists of a comprehensive set of proprietary technologies and capabilities, including the use of advanced genetic engineering and plant cell culture technology, enabling us to produce complex, proprietary, and biologically equivalent proteins for a variety of human diseases. Our protein expression system facilitates the creation and selection of high-expressing, genetically-stable cell lines capable of expressing recombinant proteins.
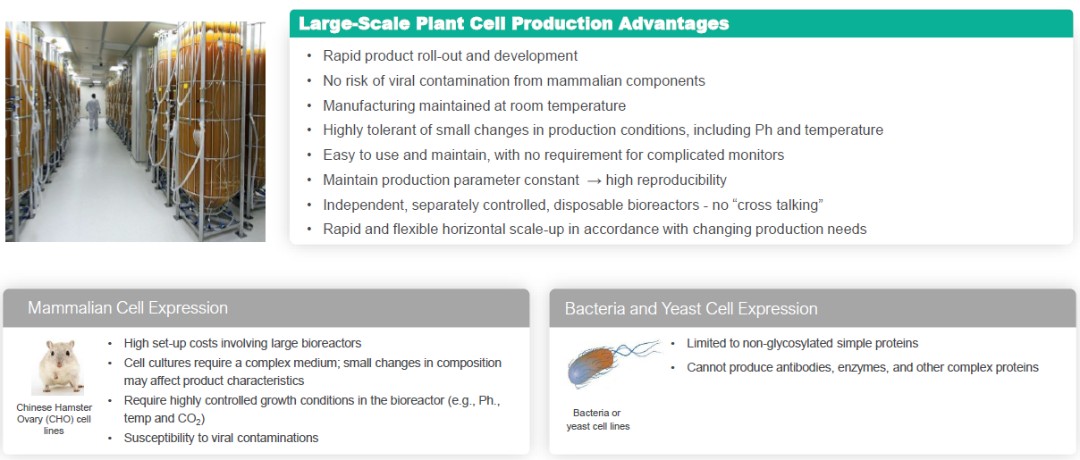
Our ProCellEx technology allows for many unique advantages, including: biologic optimization; an ability to handle complex protein expressions; flexible manufacturing with improvements through efficiencies, enhancements and/or rapid horizontal scale-ups; a simplified production process; elimination of the risk of viral contaminations from mammalian components; and intellectual property advantages.
We are the first and only company to gain FDA approval of a protein produced through plant cell-based expression, and with the recent approval of Elfabrio, we now have two FDA approved proteins produced through our platform. Our ProCellEx platform uses flexible polyethylene disposable bioreactors and is optimized for plant cell cultures. As opposed to the large stainless-steel bioreactors commonly used for recombinant protein production, our ProCellEx bioreactors are easy to use and maintain and allow for the major advantage of rapid horizontal scale-up.
Plant Cell Production Advantages
19
ProCellEx®: Protalix’s Differentiated Plant Cell Protein Expression Platform
Products and Product Pipeline
Elfabrio (PRX-102) for the Treatment of Fabry Disease
On May 5, 2023, the EC announced that it had approved the MAA for Elfabrio and on May 9, 2023, the FDA announced that it had approved the BLA for Elfabrio, each for adult patients with Fabry disease. Both approvals cover the 1 mg/kg every two weeks dosage. The EMA approval followed the February 2023 adoption of a positive opinion and recommendation of marketing authorization for Elfabrio by the CHMP. Elfabrio was approved by the FDA with a boxed warning for hypersensitivity reactions/anaphylaxis, consistent with ERT class labeling, and Warnings/Precautions providing guidance on the signs and symptoms of hypersensitivity and infusion-associated reactions seen in the clinical studies as well as treatments to manage such events should they occur. The Warnings/Precautions for MPGN alert prescribers to the possibility of MPGN and provide guidance for appropriate patient management. Overall, the FDA review team concluded that in the context of Fabry disease as a rare, serious disease with limited therapeutic options that may not be suitable to all individual patients, the benefit-risk of Elfabrio is favorable for the treatment of adults with confirmed Fabry disease.
In August and September of 2023, Elfabrio was approved in Great Britain and Switzerland, respectively, for long-term enzyme replacement therapy in adult patients with a confirmed diagnosis of Fabry disease.
20
Elfabrio, an ERT, is our proprietary, plant cell culture expressed enzyme, and a chemically modified stabilized version of, the recombinant α-Galactosidase-A protein, a lysosomal enzyme. Fabry disease is a serious life-threatening rare genetic disorder. Fabry patients lack or have low levels of α-galactosidase-A resulting in the progressive accumulation of abnormal deposits of a fatty substance called globotriaosylceramide, or Gb3, in blood vessel walls throughout their body. The ultimate consequences of Gb3 deposition range from episodes of pain and impaired peripheral sensation to end-organ failure, particularly of the kidneys, but also of the heart and the cerebrovascular system. Fabry disease occurs in one person per 40,000 to 60,000 males.
The global market for Fabry disease, that includes Sanofi’s Fabrazyme®, Shire’s (acquired by Takeda Pharmaceutical Company Limited) Replagal®, and Amicus Therapeutics’ Galafold®, among others, was approximately $2.0 billion in 2022, is forecasted to be approximately $2.0 billion in 2023 and is forecasted to grow at a CAGR of approximately 10% from 2022-2029.
In preclinical studies, PRX-102 showed significantly longer half-life due to higher enzyme stability, enhanced activity in Fabry disease affected organs leading to reduction of the accumulated substrate and reduced immunogenicity, which together can potentially lead to improved efficacy through increased substrate clearance and significantly lower formation of anti-drug antibodies, or ADAs. In 2015, we completed a phase I/II clinical trial of PRX-102 (NCT01678898), which was a worldwide, multi-center, open-label, dose ranging study designed to evaluate the safety, tolerability, pharmacokinetics, immunogenicity and efficacy parameters of PRX-102 in adult patients with Fabry disease. Our phase III clinical program, which has now been completed, included the following three separate studies:
The BALANCE study (PB-102-F20, NCT02795676), a pivotal 24-month, randomized, double blind, active control study of PRX-102 in adult Fabry patients with deteriorating renal function that was designed to evaluate the safety and efficacy of 1 mg/kg of PRX-102 administered every two weeks compared to agalsidase beta;
The BRIDGE study (PB-102-F30, NCT03018730), a 12-month open-label, single arm switch-over study evaluating the safety and efficacy of PRX-102, 1 mg/kg infused every two weeks, in up to 22 Fabry patients previously treated with agalsidase alfa for at least two years and on a stable dose for at least six months; and
The BRIGHT study (PB-102-F50, NCT03180840), a multicenter, multinational open-label, switch-over study designed to evaluate the safety, efficacy and pharmacokinetics of treatment with 2 mg/kg of PRX-102 administered every four weeks for 52 weeks (a total of 14 infusions). The 2 mg/kg every four weeks dosage was not approved by the EMA, FDA or any other jurisdiction.
Patients who completed the BALANCE, BRIDGE and BRIGHT studies, and the extension of the phase I/II study, were offered the opportunity to continue PRX-102 treatment in one of two long-term open-label extension studies. Overall, 126 subjects who participated in our PRX-102 clinical program initially opted, with the advice of the treating physician, to enroll in one of our long-term, open label, extension studies of PRX-102: 97 patients in the 1 mg/kg every two weeks extension study (PB-102-F60, NCT03566017) (10 subjects who completed an extension study from the phase I/II study, 18 subjects who completed the BRIDGE study, 69 subjects who completed the BALANCE study); and 29 subjects who completed the BRIGHT study in the 2 mg/kg every four weeks extension study (PB-102-F51, NCT03614234). Two of the subjects in the PB-102-F51 study are being treated with 1 mg/kg every two weeks. As of March 1, 2023, sponsorship of the two extension studies was transferred to Chiesi, and Chiesi is now administering the open-label extension studies.
Over time, and as Elfabrio is approved for marketing in different jurisdictions, participants withdraw from the open-label extension studies. Some of the withdrawals transfer to a commercial setting, others withdraw for other reasons.
A BLA for Elfabrio for the treatment of adult patients with Fabry disease was first submitted to the FDA on May 27, 2020 under the FDA’s Accelerated Approval pathway, but resulted in a CRL. The BLA was resubmitted to the FDA on November 9, 2022.
The MAA was submitted to the EMA on February 7, 2022, after the October 8, 2021 meeting we held, together with Chiesi, with the Rapporteur and Co-Rapporteur of the EMA regarding PRX-102.
The FDA publicly released the internal review documents for Elfabrio (pegunigalsidase alfa-iwxj) injection BLA 761161. These documents provide previously unavailable additional information regarding the basis for the FDA’s May 2023 approval decision. In particular, the FDA determined that substantial evidence of effectiveness for Elfabrio in Fabry patients was established with one adequate and well-controlled study (Study PB-102-F01/02) with confirmatory evidence provided by the BALANCE study (also referred to as Study PB-102-F20). The FDA review team also concluded that the BALANCE study met its primary efficacy endpoint, which assessed the annualized rate of change in eGFR (estimated glomerular filtration rate) over 104 weeks (two years).
21
However, the FDA also determined that the results from the BALANCE study did not support a non-inferiority claim to the comparator product due to the lack of data to support a non-inferiority margin.
In February 2020, we, together with Chiesi, announced an agreement with the FDA for the Initial Pediatric Study Plan (iPSP) for PRX-102, which is intended to be initiated post-marketing approval. The joint announcement was made after completion of discussions with the FDA and receipt of confirmation from the FDA in an official “Agreement Letter” which outlines an agreed-upon approach to evaluate the safety and efficacy of PRX-102 in pediatric Fabry patients in a clinical trial to be performed by Chiesi with our collaborative efforts.
Chiesi, together with Protalix, participated in an Oral Explanation at a meeting of the EMA’s Committee for Orphan Medicinal Products (COMP) held on March 21, 2023, as part of the Orphan Drug Designation maintenance process. Following the meeting, Chiesi formally withdrew the application for Orphan Drug Designation for PRX-102. The EC first granted Orphan Drug Designation for PRX-102 for the treatment of Fabry disease in December 2017.
Phase III BALANCE Study
The Clinical Study Report for the BALANCE study was completed in July 2022. The final analysis confirmed the positive topline results (announced in April 2022) and favorable tolerability profile. A total of 78 patients who were previously treated with agalsidase beta for at least one year with an eGFR slope at screening worse than -2 mL/min/1.73 m2/year were enrolled in the study. Patients were randomized on a 2:1 ratio for switching to PRX-102 or continuing on agalsidase beta. A total of 77 patients were treated; 52 with PRX-102 and 25 with agalsidase beta. Approximately 40% of the enrolled patients were female.
The primary endpoint of the BALANCE study is the comparison in the annualized rate of decline of eGFR slope between the agalsidase beta and PRX-102 treatment arms. eGFR is considered a reliable and accepted test to measure kidney function and stage of kidney disease. Additional parameters evaluated include: cardiac assessment, lyso-Gb3 (a biomarker for monitoring Fabry patients during therapy), pain, quality of life, immunogenicity, Fabry Clinical Events, pharmacokinetics and other parameters.
Based on the original primary analysis (random intercept random slope (RIRS)), the estimated mean eGFR slopes were -2.4 mL/min/1.73 m2 /year and -2.3 mL/min/1.73 m2 /year in the PRX-102 and agalsidase beta arms, respectively, and the treatment difference was -0.1 (95% CI: -2.2, 2.1) mL/min/1.73 m2 /year. Based on the ANCOVA adjusted for continuous baseline proteinuria, the estimated mean eGFR slopes were -2.0 and -3.1 mL/min/1.73 m2 /year in the PRX-102 and agalsidase beta arms, respectively, and the treatment difference was 1.1 (95% CI: -0.8, 3.1) mL/min/1.73 m2 /year. Based on quantile regression model, the median of the eGFR slope in the PRX-102 arm was -2.514 mL/min/1.73 m2/year (95% CI:-3.788, -1.240) and -2.155 mL/min/1.73 m2/year (95% CI:-3.805, -0.505) in the agalsidase beta arm, demonstrating a large overlap in the confidence intervals of the two arms. The difference in medians (95% confidence interval) is -0.359 mL/min/1.73 m2/year (-2.444, 1.726). Additional sensitivity and supportive analyses investigated mean eGFR slopes using other statistical models. These models yielded results similar to the primary analysis and confirming the comparability between the treatment arms. These results supported the robustness of the methodology used for comparisons of treatment effects in the BALANCE study. The results of the analyses on eGFR slopes were further supported by the analysis of change from baseline in the average eGFR at the last two visits (100 and 104 weeks). The estimated mean changes were -3.0 and -3.8 mL/min/1.73 m2 in the PRX102 and agalsidase beta arms, respectively. The difference in mean change (PRX-102 – agalsidase beta) was 0.8 (95% CI: -3.0, 4.6) mL/min/1.73 m2 or annualized change of 0.4 (95% CI: -1.5, 2.3) mL/min/1.73 m2/year.
The study population (ITT analysis set) was composed of 47 males (61.0%) and 30 females (39.0%), with a mean (range) age of 44.3 (18-60) years. The mean duration of prior treatment with agalsidase beta was approximately six years. At baseline, mean (SD) eGFR was 73.69 ml/min/1.73 m2 (20.32) and median eGFR was 74.51 ml/min/1.73 m2; mean (SD) eGFR slope was -8.10 mL/min/1.73 m2/year (5.92) and median eGFR slope was -7.25 ml/min/1.73 m2/year.
A comparable efficacy response was also observed across biomarkers and functional systems relevant to Fabry disease, as demonstrated via secondary endpoints, where in some cases the trend was in favor of PRX-102 and in some in favor of agalsidase beta, but the actual difference between the two arms is always clinically small, supporting the comparability of the two treatments.
Key secondary endpoints included Urine protein creatinine ratio (UPCR) as indicator of proteinuria, plasma levels of lyso–Gb3, imaging marker of cardiac remodeling (Left Ventricular Mass Index, LVMI, by cardiac MRI), disease severity (by Mainz Severity Score Index, MSSI), pain severity (Short Form Brief Pain Inventory, BPI) and quality of life (EQ-5D-5L). Both treatments showed either a stabilization of clinical parameters (e.g., for eGFR, eGFR slope and UPCR) or prevention of further progression of Fabry disease (e.g., LVMI, MSSI).
22
For an overview of primary and secondary endpoints collected in the BALANCE study, please refer to the Table 1 below.
Table 1: Summary Table of Comparison of Treatment Benefit Data in the BALANCE Study, (Mean (SE) [median]), Efficacy Population
Parameter | PRX-102 (N = 52) | Agalsidase beta (N = 25) | |||
eGFR (ml/min/1.73 m2) | n | n | |||
Baseline | 52 | 73.46 (2.80) [73.45] | 25 | 74.16 (4.19) [74.85] | |
Month 24 | 47 | 70.53 (3.19) [69.35] | 24 | 72.05 (4.69) [74.48] | |
Change from Baseline | 47 | -3.60 (1.58) [-2.39] | 24 | -1.97 (1.51) [-3.20] | |
eGFR slope (ml/min/1.73 m2/yr) | Baseline | 52 | -8.03 (0.92) [-6.70] | 25 | -8.25 (0.85) [-7.84] |
Month 24 | 51 | -2.38 (1.25) [-2.51] | 25 | -2.31 (0.71) [-2.16] Q1; Q3: -4.6; -0.5 | |
Reaching kidney therapeutic goala | Month 24 | 52 | 41 patients (80.4%) | 25 | 20 patients (80.0%) |
UPCR | Baseline | 52 | 0.441 (0.084) | 25 | 0.284 (0.097) |
Month 24 | 45 | 0.480 (0.118) | 24 | 0.489 (0.162) | |
Change from Baseline | 45 | 0.088 (0.067) | 24 | 0.197 (0.085) | |
Plasma lyso-Gb3 (nM) | Baseline | 52 | 26.22 (3.78) [15.20] | 25 | 32.14 (7.08) [17.60] |
Month 24 | 46 | 29.22 (4.48) [18.80] | 22 | 19.65 (3.60) [15.30] | |
Change from Baseline | 46 | 3.30 (1.38) [1.15] | 22 | -8.74 (4.85) [-1.50] | |
LVMI (g/m2) | Baseline | 40 | 75.97 (5.13) | 22 | 82.22 (6.34) |
Month 24 | 35 | 71.56 (5.20) | 20 | 82.43 (8.39) | |
Change from Baseline | 28 | -0.64 (2.69) | 19 | 0.29 (3.73) | |
23
MSSI (overall score)a | Baseline | 49 | 23.18 (1.42) | 25 | 25.16 (2.14) |
Month 24 | 46 | 22.11 (1.80) | 23 | 27.09 (2.30) | |
Change from Baseline | 44 | -2.07 (0.77) | 23 | 2.04 (1.10) | |
BPI (score for pain at its worst)b | Baseline | 52 | 3.5 (0.4) | 25 | 2.6 (0.6) |
Month 24 | 45 | 3.3 (0.5) | 22 | 3.0 (0.7) | |
Change from Baseline | 45 | -0.1 (0.5) | 22 | 0.6 (0.6) |
BPI=brief pain inventory; eGFR=estimated glomerular filtration rate; lyso-Gb3=globotriaosylsphingosine; LVMI=Left Ventricular Mass Index; MSSI=Mainz Severity Score Index; UPCR=Urine Protein Creatinine Ratio.
a Wanner 2018; b Higher scores indicate higher symptom severity.
Forty-seven (90.4%) patients in the PRX-102 arm experienced at least one treatment-emergent adverse event (TEAE) compared to 24 (96.0%) in the agalsidase beta arm. The number of events adjusted to 100 years of exposure is 572.36 events for the PRX-102 arm and 816.85 events for the agalsidase beta arm.
Treatment-related adverse events were reported for 21 (40.4%) patients in the PRX-102 arm compared to 11 (44.0%) in the agalsidase beta arm. The number of treatment-related events adjusted to 100 years of exposure is 42.85 events for the PRX-102 arm and 152.91 events for the agalsidase beta arm.
Usage of infusion pre-medication was tapered down during the study, if possible, for all patients. At baseline, 21 (40.4%) patients in the PRX-102 arm used infusion premedication compared to 16 (64.0%) in the agalsidase beta arm. At the end of the study, only three out of 47 (6.4%) patients in the PRX-102 arm used infusion premedication compared to three out of 24 (12.5%) in the agalsidase beta arm. Even with this reduction in use of premedication, there were fewer reported infusion-related reactions with PRX-102: 11 (21.2%) patients in the PRX-102 arm experienced a total of 13 events compared to six (24.0%) patients experiencing a total of 51 events in the agalsidase beta arm. The number of infusion-related reactions adjusted to 100 infusions is 0.5 for the PRX-102 arm and 3.9 for the agalsidase beta arm.
Assessment of immunogenicity, that is, the existence and development of anti PRX-102 antibodies or anti-agalsidase beta antibodies, in the study indicated that for the PRX-102 arm, 18 (34.6%) patients were ADA positive at baseline, of which 17 (94.4%) had neutralizing antibody activity. For the agalsidase beta arm, eight (32.0%) patients were ADA positive at baseline, of which seven (87.5%) had neutralizing antibody activity. Only a small number of patients showed treatment-emergent ADA. At the end of the two-year study, 11 (23.4%) patients who received PRX-102 were ADA positive, of which seven (63.6%) had neutralizing antibody activity, while in the agalsidase beta arm six (26.1%) were ADA-positive and all six (100%) had neutralizing antibody activity. There was little change in the percentage of patients who were ADA positive, with a trend of reduction observed in the PRX-102 arm and stability in the agalsidase beta arm. The proportion of patients with neutralizing ADA decreased in the PRX-102 arm while it remained stable in the agalsidase beta arm.
Out of the 78 randomized patients, six patients discontinued the study: out of the five (9.4%) from the PRX-102 arm, one patient withdrew consent prior to the first infusion, two discontinued due to personal reasons, and two due to adverse events (one due to an unrelated adverse event and one due to a treatment related adverse event); one (4%) patient from the agalsidase beta arm discontinued for personal reasons. There were no deaths in this study.
Considering that in the trial, patients in the PRX-102 arm were exposed for the first time to the novel enzyme, tolerability data appear favorable for PRX-102 and in line with what was observed in the previous clinical studies of PRX-102.
Of the patients who completed the trial from both the PRX-102 and agalsidase beta treatment arms, 69 have opted, with the advice of the treating physician, to receive PRX-102 1 mg/kg every two weeks in the long-term open-label extension study which is now sponsored by Chiesi.
The results of the direct, blinded comparison of PRX-102 to agalsidase beta, for the primary efficacy renal endpoints (i.e., eGFR change, eGFR slope) and for the main secondary endpoints (e.g., urine protein to creatinine ratio [UPCR] LVMI, MSSI, BPI) strongly suggest comparability in treatment effects between the two treatments.
At the same time a potentially favorable safety profile was identified based on lower rates of IRR, lower ADA positivity, and less premedication use in the PRX-102 arm compared to agalsidase beta. Overall, a positive benefit-risk balance was confirmed.
24
Phase III BRIDGE Study
The BRIDGE study was completed in December 2019. In the study, patients were screened and evaluated over three months while continuing agalsidase alfa treatment.
Final results of the data generated in the BRIDGE study showed substantial improvement in renal function as measured by mean annualized eGFR slope in both male and female patients. Twenty of 22 patients completed the 12-month treatment duration. Eighteen of the patients who completed the study opted to roll over to a long-term extension study and continue to be treated with PRX-102. In the study, the mean annualized eGFR slope of the study participants improved from -5.90 mL/min/1.73 m2/year while on agalsidase alfa to -1.19 mL/min/1.73 m2/year on PRX-102 in all patients. Male patients improved from -6.36 mL/min/1.73 m2/year to -1.73 mL/min/1.73 m2/year and female patients improved from -5.03 mL/min/1.73 m2/year to -0.21 mL/min/1.73 m2/year. Following the switch to PRX-102, there was a decrease in patients with progressing or fast progressing kidney disease which is consistent with the therapeutic goals for Fabry disease, as identified by Christoph Wanner, et. al., in 2019, and most patients achieved a stable status post-switch.
PRX-102 was well-tolerated in the BRIDGE study, with all adverse events being transient in nature without sequelae. Of the 22 patients enrolled in the BRIDGE study, the majority of TEAEs were mild or moderate in severity, with two patients (9.1%) withdrawing from the therapy due to hypersensitivity reaction that was resolved. The most common moderate TEAEs were nasopharyngitis, headache and dyspnea.
An immunogenicity assessment indicated that four out of 20 patients (20%) developed persistent ADAs over the course of the study, of which two had neutralizing activity.
Baseline characteristics of the 20 patients who completed the study, ranging from ages 28 to 60 years, were as follows: mean eGFR 75.87 mL/min/1.73 m2 in males, and 86.14 mL/min/1.73 m2 in females and plasma lyso-Gb3 were 51.81 nM and 13.81 nM in males and females, respectively. While lyso-Gb3 levels remain slightly high, particularly within the male cohort, continuous reduction in lyso-Gb3 levels was observed of 19.55 nM (32.35%) in males and 4.57 nM (29.81%) in females.
Of the patients who completed the trial, 18 have opted, with the advice of the treating physician, to continue receiving PRX-102 1 mg/kg every two weeks in a long-term open-label extension study which now sponsored by Chiesi.
Phase III BRIGHT Study
The BRIGHT study, which studied the 2 mg/kg every four weeks dosage, was completed in June 2020. This dosage was not approved by the EMA or the FDA.
Enrollment in the study included 30 adult patients (24 males and 6 females) with mean (SD) age of 40.5 (11.3) years, ranging from 19 to 58 years previously treated with a commercially available ERT (agalsidase beta or agalsidase alfa), for at least three years and on a stable dose administered every two weeks. To determine eligibility for participation in the study, candidates were screened to identify and select Fabry patients with clinically stable kidney disease. The most common Fabry disease symptoms at baseline were acroparesthesia, heat intolerance, angiokeratomas and hypohydrosis. Patients who matched the criteria were enrolled in the study and switched from their current treatment of IV infusions every two weeks to 2 mg/kg of PRX-102 every four weeks for 12 months. Patients participating in the study were evaluated, among other disease parameters, to determine if their kidney disease had not further deteriorated while being treated with the four-week dosing regimen as measured by eGFR and for lyso-Gb3 levels as a Fabry biomarker, as well as other parameters. In addition, participating patients were evaluated to assess the safety and tolerability of PRX-102.
We announced final results from the BRIGHT study in March 2022. The results indicate that 2 mg/kg of PRX-102 administered by intravenous infusion every four weeks was well tolerated, and Fabry disease assessed by eGFR slope and plasma lyso-Gb3 was stable throughout PRX-102 treatment in adult Fabry patients. None of the patients without ADAs at screening developed treatment-induced ADAs following the switch to PRX-102 treatment.
All 30 patients received at least one dose of PRX-102, and 29 patients completed the one-year study. Of these 29 patients, 28 received the intended regimen of 2 mg/kg every four weeks throughout the entire study, while one patient was switched to 1 mg/kg PRX-102 every two weeks per protocol at the 11th infusion. One patient withdrew from the study after the first infusion due to a traffic accident.
First infusions of PRX-102 were administered under controlled conditions at the investigation site. Based on the protocol-specified criteria, patients were able to receive their PRX-102 infusions at a home care setup once the applicable Investigator and Sponsor
25
Medical Monitor agreed that it was safe to do so. Safety and efficacy exploratory endpoints were assessed throughout the 52-week study.
Overall, 33 of 183 total TEAEs reported in nine (30.0%) of the patients were considered treatment related; all were mild or moderate in severity and the majority were resolved at the end of the study. There were no serious or severe treatment-related TEAEs and no TEAEs led to death or study withdrawal. Of the treatment-related TEAEs, 27 were infusion-related reactions (IRRs) and the remainder were single events of diarrhea, erythema, fatigue, influenza-like illness, increases urine protein/creatinine ratio, and urine positive for white blood cells. The 27 IRRs were reported in five (16.7%) patients, all male. All IRRs occurred during the infusion or within two hours post-infusion; no events were recorded between two and 24 hours post-infusion.
Study outcome measures show that plasma lyso-Gb3 concentrations remained stable during the study with a mean change (±SE) of 3.01 nM (0.94) from baseline (19.36 nM ±3.35) to Week 52 (22.23 ±3.60 nM). Mean absolute eGFR values were stable during the 52-week treatment period, with a mean change from baseline of -1.27 mL/min/1.73 m2 (1.39). Mean (SE) eGFR slope, at the end of the study, for the overall population, was -2.92 (1.05) mL/min/1.73 m2/year indicating stability.
Following a survey of participants using the Quality of Life EQ-5D-5L questionnaire, responses indicate that patient perception of their own health remained high and stable throughout the 52-week study duration, with overall health mean (SE) scores of 78.3 (3.1) and 82.1 (2.9) at baseline and Week 52, respectively, in a 0 to 100 scale. Using the short-form Brief Pain Inventory, or questionnaire, approximately 75% of study participants had an improvement or no change in average pain severity at Week 52 (compared to baseline). The short-form BPI interference items also remained stable during the study. Pain-related results indicate that there was no increase and/or relapse in pain. No Fabry clinical events were reported during the study.
Phase I/II Study
Sixteen adult, naïve Fabry patients (9 male and 7 female) completed our phase I/II clinical trial of PRX-102, each in one of three dosing groups, 0.2 mg/kg, 1 mg/kg and 2 mg/kg. Each patient received IV infusions of PRX-102 every two weeks for 12 weeks, with efficacy follow-up after six-month and twelve-month periods. A majority of the patients who completed the trial opted to continue receiving PRX-102 in an open-label, 60-month extension study under which all patients were switched to receive 1 mg/kg of the drug, the selected dose for our BALANCE and BRIDGE studies.
The adult symptomatic, ERT-naïve Fabry disease patients enrolled in the phase I/II study were evaluated for Gb3 levels in kidney biopsies and for plasma lyso-Gb3 concentration by the quantitative BLISS methodology. Biopsies were available from 14 patients. The outcome of ≥ 50% reduction in the average number of Gb3 inclusions per kidney PTC from baseline to Month 6 was demonstrated in 11 of 14 (78.6%) of the patients treated with PRX-102. The overall results demonstrate that PRX-102 reaches the affected tissue and reduces kidney Gb3 inclusions burden and lyso-Gb3 in the circulation. A high correlation was found between the two Fabry disease biomarkers, reduction of kidney Gb3 inclusions and the reduction of plasma lyso-Gb3 over six months of treatment.
Data was recorded at 24 months from 11 patients who completed 12 months of the long-term open-label extension trial that succeeded the phase I/II study. Patients who did not continue in the extension trial included: female patients who became or planned to become pregnant and therefore were unable to continue in accordance with the study protocol; and patients who relocated to a location where treatment was not available under the clinical study.
Results show that lyso-Gb3 levels decreased approximately 90% from baseline. Renal function remained stable with mean eGFR levels of 108.02 and 107.20 at baseline and 24 months, respectively, with a modest annual eGFR slope of -2.1. An improvement across all the gastrointestinal symptoms evaluated, including severity and frequency of abdominal pain and frequency of diarrhea, was noted. Cardiac parameters, including LVM, LVMI and EF, remained stable with no cardiac fibrosis development detected. In conclusion, an improvement of over 40% in disease severity was shown as measured by the Mainz Severity Score Index, or MSSI, a score compiling the different elements of the disease severity including neurological, renal and cardiovascular parameters. In addition, an improvement was noted in each of the individual parameters of the MSSI.
The majority of adverse events were mild-to-moderate in severity, and transient in nature. During the first 12 months of treatment, only three of 16 patients (less than 19%) formed ADAs of which two of these patients (less than 13%) had neutralizing antibodies. Importantly, however, the ADAs turned negative for all three of these patients following 12 months of treatment. The ADA positivity effect had no observed impact on the safety, efficacy or continuous biomarker reduction of PRX-102.
Commercialization Agreements with Chiesi Farmaceutici
We have entered into two exclusive global licensing and supply agreements for PRX-102 for the treatment of Fabry disease with Chiesi. The agreements have significant revenue potential for Protalix. Under the agreements, Protalix Ltd. has received $50.0 million
26
in upfront payments and development cost reimbursements of $45.0 million, and is entitled to approximately $1.0 billion in potential milestone payments as well as additional payments as consideration for drug product supply. The additional payments for drug product supplied are based on the average selling price of the drug product in the relevant territory multiplied by tiered payments, as detailed below. During the quarter ended June 30, 2023, we received net proceeds of $20.0 million representing a milestone payment earned upon the FDA’s approval of Elfabrio for adult Fabry patients.
On October 19, 2017, Protalix Ltd. and Chiesi entered into the Chiesi Ex-US Agreement pursuant to which Chiesi was granted an exclusive license for all markets outside of the United States to commercialize PRX-102. Under the Chiesi Ex-US Agreement, Chiesi made an upfront payment to Protalix Ltd. of $25.0 million in connection with the execution of the agreement, and Protalix Ltd. was entitled to additional payments of up to $25.0 million in development costs in the aggregate, capped at $10.0 million per year. Protalix Ltd. is also eligible to receive additional payments of up to a maximum of $320.0 million in regulatory and commercial milestone payments. Protalix Ltd. agreed to manufacture all of the PRX-102 needed for all purposes under the agreement, subject to certain exceptions, and Chiesi will purchase PRX-102 from Protalix Ltd., subject to certain terms and conditions. Chiesi is required to make tiered payments for drug product purchased from Protalix based on the average selling price of the drug product in the relevant territory multiplied by 15% to 35%, depending on the amount of annual net sales outside of the United States.
On July 23, 2018, Protalix Ltd. entered into the Chiesi US Agreement with respect to the development and commercialization of PRX-102 in the United States. Protalix Ltd. received an upfront, non-refundable, non-creditable payment of $25.0 million from Chiesi and was entitled to additional payments of up to a maximum of $20.0 million to cover development costs for PRX-102, capped at $7.5 million per year. Protalix Ltd. is also eligible to receive additional payments of up to a maximum of $760.0 million, in the aggregate, in regulatory and commercial milestone payments. Chiesi agreed to make tiered payments for drug product purchased from Protalix based on the average selling price of the drug product in the United States multiplied by 15% to 40%, subject to certain terms and conditions, depending on the amount of annual net sales in the United States.
On May 13, 2021, we signed a binding term sheet with Chiesi amending the Chiesi Agreements in order to provide our company with near-term capital. Chiesi agreed to make a $10.0 million payment to us before the end of the second quarter of 2021 in exchange for a $25.0 million reduction in a longer term regulatory milestone payment provided in the Chiesi EX-US Agreement. All other regulatory and commercial milestone payments remain unchanged. We received the payment in June 2021. We also agreed to negotiate certain manufacturing related matters.
On August 29, 2022, we entered into the F/F Agreement and the Letter Agreement with Chiesi. Under the F/F Agreement, we agreed to supply Chiesi with drug substance for PRX-102 and, following relevant technology and technical information transfer activities, Chiesi has agreed, among other things, to provide us with commercial fill/finish services for PRX-102, including to support the anticipated global launch of PRX-102. The F/F Agreement will expire December 31, 2025, unless terminated earlier in accordance with its terms and may be extended by mutual agreement in writing for an additional period of seven years. The Letter Agreement changed our obligations and those of Chiesi under the License Agreements with respect to, among other things, the evaluation, selection and establishment of an initial alternate source of commercial fill/finish services for PRX-102. In addition, the Letter Agreement amended certain provisions of the License Agreements to reflect the appointment of Chiesi as a supplier to our company of commercial fill/finish services and the potential establishment of an initial alternate source of commercial fill/finish services.
As of March 1, 2023, sponsorship of the two extension studies was transferred to Chiesi, and Chiesi is now administering the extension studies.
Elelyso® for the Treatment of Gaucher Disease
Elelyso (taliglucerase alfa), our first commercial product, was approved by the FDA in 2012 for injection as an enzyme replacement therapy (ERT) for the long-term treatment of adult patients with a confirmed diagnosis of type 1 Gaucher disease. In August 2014, the FDA approved Elelyso for injection for pediatric patients. Elelyso is the first plant cell derived recombinant protein to be approved by the FDA for the treatment of Gaucher disease and is now approved in over 20 markets.
Gaucher disease, also known as glucocerebrosidase, or GCD, deficiency, is a rare genetic autosomal recessive disorder and one of the most common Lysosomal Storage Disorders, or LSDs, in the world. It is one of a group of disorders that affect specific enzymes that normally break down fatty substances for reuse in the cells. If the enzymes are missing or do not work properly, the substances can build up and become toxic. Gaucher disease occurs when a lipid called glucosylceramide accumulates in the cells of the bone marrow, lungs, spleen and liver, and sometimes the brain. Gaucher disease symptoms can include fatigue, anemia, easy bruising and bleeding, severe bone pain and easily broken bones, and distended stomach due to an enlarged spleen and thrombocytopenia. Epidemiology of Gaucher disease varies; recent literature provides that prevalence of Gaucher disease ranges from 0.70 to 1.75 per 100,000 in the
27
general population. In people of Ashkenazi Jewish heritage, estimates of occurrence vary from approximately 1 in 400 to 1 in 850 people.
The global market for Gaucher disease, that includes Sanofi’s Cerezyme®, Shire’s Vpriv® and Sanofi’s Cerdelga®, among others, was $1.6 billion in 2022, is forecasted to be approximately $1.4 billion in 2023 and is forecasted to grow at a CAGR of approximately 1.6% from 2023-2029.
Commercialization Agreements for Elelyso
We have licensed to Pfizer the global rights to Elelyso in all markets excluding Brazil. Pfizer retains 100% of revenue and reimburses 100% of direct costs. We manufacture drug substance for Pfizer, subject to certain terms and conditions.
For the first 10-year period after the execution of the October 2015 Amended Pfizer Agreement, we have agreed to sell drug substance to Pfizer for the production of Elelyso, and Pfizer maintains the right to extend the supply period for up to two additional 30-month periods subject to certain terms and conditions. In a subsequent amendment, we agreed that after the completion of the first 10-year supply period, the supply term would automatically extend for a five-year period (i.e., until October 2030).
We maintain distribution rights to Elelyso in Brazil through a supply and technology transfer agreement with Fiocruz, an arm of the Brazilian MoH.
Uricase (PRX-115)
PRX-115 is our plant cell-expressed recombinant PEGylated uricase (urate oxidase) – a chemically modified enzyme under development for the potential treatment of severe gout. We use ProCellEx to express an optimized recombinant uricase enzyme under development for the potential treatment of severe gout which we are designing to lower uric acid levels while having low immunogenicity and increased half-life in the circulation. Pre-clinical data demonstrates stable PK profile and long half-life, low immunogenic risk and high specific activity which supports the potential of PRX-115 to be a safe and effective treatment for severe gout. Results from the one-month multiple dosing toxicity studies in two species demonstrate that PRX-115 is well tolerated.
On March 21, 2023, the first patient was dosed in our phase I First in Human (FIH) clinical trial of PRX-115, a double-blind, placebo-controlled trial designed to evaluate the safety, pharmacokinetics, pharmacodynamics (reduction of uric acid) and immunogenicity of PRX-115 in patients with elevated uric acid levels (>6.0 mg/dL). The trial is a single ascending dose (SAD) study with up to seven cohorts, and patients are to be randomized 3:1 to receive a single intravenous (IV) dose of PRX-115 or a placebo. The study is being conducted at New Zealand Clinical Research (NZCR) under the New Zealand Medicines and Medical Devices Safety Authority (MedSafe) and the Health and Disability Ethics Committee (HDEC) guidelines, and is expected to enroll up to 56 patients with no previous exposure to PEGylated uricase. As of September 30, 2023, 32 patients have been dosed in this trial.
Gout is the most common inflammatory arthritis in the United States, affecting an estimated 9.2 million adults. Gout is caused by factors that elevate serum uric acid, or sUA, levels, which may include diet or genetic predisposition and environmental factors leading to hyperuricemia and tissue deposition of monosodium urate crystals, tophi, in joints and soft tissues, causing acute and chronic inflammation, and is characterized by recurrent flares. Gout leads to substantial morbidity by causing severe pain, reduced quality of life, decreased physical function, increased healthcare costs, and lost economic productivity. Furthermore, gout is strongly associated with metabolic syndrome, and may contribute to myocardial infarction, type 2 diabetes mellitus, chronic kidney disease, or CKD, and premature mortality.
Severe gout is generally described as a state of gout in which there is a presence of monosodium urate crystals with any of the following: frequent recurrent gout flares, chronic gouty arthritis, subcutaneous tophi or disease elements of gout seen via imaging. It is estimated that approximately 2% of the gout patient population is considered to have chronic refractory disease, and we believe the incidence of severe gout is higher.
Currently available urate-lowering therapies, or ULTs, can be effective in treating gout. However, we believe that new effective, safe therapies are needed to treat severe gout and chronic refractory gout regardless of treatment history. One treatment option may be a therapeutic use of the uricase enzyme which converts uric acid to allantoin, which is easily eliminated through urine. The uricase enzyme does not exist naturally in humans. To date, two variants of recombinant uricases are approved for marketing: (i) Krystexxa® (pegloticase) for treatment of chronic gout refractory to conventional therapy (gout patients who have contraindication/failure of other lowering uric acid treatments) and (ii) Elitek®, indicated for the treatment of tumor lysis syndrome but not gout. Both have a black box warning for anaphylaxis and other major side-effects. In particular, 89% of patients treated with Krystexxa developed an immunogenic response associated with a failure to maintain normalization of serum uric acid levels over a 6-month therapy cycle. In addition, a phase IV study demonstrates that co-treatment with Krystexxa and methotrexate prolongs efficacy and increases
28
tolerability in patients with refractory gout. Krystexxa is no longer marketed in the European Union. The EC withdrew the marketing authorization for Krystexxa in 2016 at the request of the marketing authorization holder which notified the EC of its decision not to market the product in the European Union for commercial reasons.
PRX-119
PRX-119 is our plant cell-expressed PEGylated recombinant human DNase I product candidate being designed to elongate half-life in the circulation for NETs-related diseases. NETs, Neutrophil extracellular traps, are web-like structures, released by activated neutrophils that trap and kill a variety of microorganisms. NETs are composed of DNA, histones, antimicrobial and pro-inflammatory proteins. Excessive formation or ineffective clearance of NETs can cause different pathological effects. NETs formation has been observed in various autoimmune, inflammatory and fibrotic conditions, diverse forms of thrombosis, cancer and metastasis. According to scientific literature, animal studies have demonstrated that DNase treatment reduces NETs toxicity. Our proprietary modified DNase I design for long and customized systemically circulating in the bloodstream, may potentially enable effective treatment of acute and chronic conditions.
Intellectual Property
A key element of our overall strategy is to establish a broad portfolio of patents to protect our proprietary technology, proprietary product and product candidates and their methods of use. As of September 30, 2023, we hold a broad portfolio of over 85 patents in Europe, the United States, Israel and additional countries worldwide, as well as over 45 pending patent applications.
Scientific Presentations and Publications
On August 21, 2023, an article disclosing six-year results from our Phase I/II clinical trial of pegunigalsidase alfa was accepted for publication by Genetics in Medicine. The article entitled “Long-term safety and efficacy of pegunigalsidase alfa: A multicenter 6-year study in adult patients with Fabry disease,” was authored by Dr. Derralynn Hughes of University College London in London, UK, a principal investigator in our clinical trials of PRX-102, and was made available online on August 24, 2023.
Yael Hayon, Ph.D., our Vice President, Research & Development, gave a presentation at the Next Generation Protein Therapeutics Summit on September 20, 2023.The summit was held at the Boston Convention and Exhibition Center, Boston MA, on September 18 through 20, 2023.
On October 21, 2023, an article entitled “Safety and efficacy of pegunigalsidase alfa in patients with Fabry disease who were previously treated with agalsidase alfa: results from BRIDGE, a phase 3 open‑label study,” was published online. The article was authored by Ales Linhart, MD, Charles University, Praha, Czech Republic, a principal investigator in our clinical trials of PRX-102, and others.
Research & Development
We continuously work on the further development of our ProCellEx plant cell expression technology and bioreactor system. In addition, we are working on the development of new products, each in different initial stages of development, for specific products for which there are unmet needs in terms of efficacy and safety. Our development strategy focuses on the utilization of different modification approaches and development improvements, customized for each protein product, in all stages of expression and development.
Critical Accounting Policies
Our significant accounting policies are more fully described in Note 1 to our consolidated financial statements appearing in this Quarterly Report. There have been no material changes to our critical accounting policies since we filed our Annual Report on Form 10-K for the year ended December 31, 2022.
The discussion and analysis of our financial condition and results of operations is based on our financial statements, which we prepared in accordance with U.S. generally accepted accounting principles. The preparation of these financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements, as well as the reported revenues and expenses during the reporting periods. As applicable to these consolidated financial statements, the most significant estimates and assumptions relate to the assessment of sales reserves and valuation allowances. On an ongoing basis, we evaluate such estimates and judgments, including those described in greater detail below. We base our estimates on historical experience and on various other factors that we believe are reasonable under
29
the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
Results of Operations
Three months ended September 30, 2023 compared to the three months ended September 30, 2022
Revenues from Selling Goods
We recorded revenues from selling goods of $10.2 million during the three months ended September 30, 2023, an increase of $1.4 million, or 16%, compared to revenues of $8.8 million for the three months ended September 30, 2022. The increase resulted primarily from an increase of $3.0 million in sales to Chiesi, following the approvals by the FDA and the EMA of Elfabrio, and of $0.6 million in sales to Brazil, partially offset by a $2.2 million decrease in sales to Pfizer.
Revenues from License and R&D Services
We recorded revenues from license and R&D services of $0.2 million for the three months ended September 30, 2023, a decrease of $5.2 million, or 96%, compared to revenues of $5.4 million for the three months ended September 30, 2022. Revenues from license and R&D services are comprised primarily of revenues we recognized in connection with the Chiesi Agreements. As of March 1, 2023, sponsorship of the extension studies was transferred to Chiesi, and Chiesi is now administering all open-label extension studies.
Cost of Goods Sold
Cost of goods sold was $4.9 million for the three months ended September 30, 2023, a decrease of $2.2 million, or 31%, from cost of goods sold of $7.1 million for the three months ended September 30, 2022. The decrease in cost of goods sold was primarily the result of the decrease in sales to Pfizer, partially offset by an increase in sales of Elfabrio to Chiesi and of Elelyso to Brazil.
Research and Development Expenses
For the three months ended September 30, 2023, our total research and development expenses were approximately $3.7 million comprised of approximately $1.0 million of subcontractor-related expenses, approximately $1.9 million of salary and related expenses, approximately $0.2 million of materials-related expenses and approximately $0.6 million of other expenses. For the three months ended September 30, 2022, our total research and development expenses were approximately $7.4 million comprised of approximately $4.9 million in subcontractor-related expenses, approximately $1.7 million of salary and related expenses, approximately $0.2 million of materials-related expenses and approximately $0.6 million of other expenses.
Total decrease in research and developments expenses for the three months ended September 30, 2023 was $3.7 million, or 50%, compared to the three months ended September 30, 2022. The decrease in research and development expenses primarily resulted from the completion of our Fabry clinical program and the regulatory processes related to the BLA and MAA review of Elfabrio by the applicable regulatory agencies.
We expect research and development expenses to continue to be our primary expense as we enter into a more advanced stage of preclinical and clinical trials for certain of our product candidates.
Selling, General and Administrative Expenses
Selling, general and administrative expenses were $3.7 million for the three months ended September 30, 2023, an increase of $0.9 million, or 32%, compared to $2.8 million for the three months ended September 30, 2022. The increase resulted primarily from an increase of approximately $0.6 million in salary and related expenses due to one-time cash bonuses and an increase in share-based compensation.
Financial Income, Net
Financial income, net were $0.2 million for the three months ended September 30, 2023, compared to financial expenses, net of $0.4 million for the three months ended September 30, 2022. The change resulted primarily from an increase of $0.3 million in interest income.
Income taxes
30
In the three months ended September 30, 2023, we recorded income taxes of approximately $0.1 million which were primarily the result of the provision for current taxes in respect of Section 174 of the TCJA.
Nine months ended September 30, 2023 compared to the nine months ended September 30, 2022
Revenues from Selling Goods
We recorded revenues from selling goods of $30.3 million during the nine months ended September 30, 2023, an increase of $9.1 million, or 43%, compared to revenues of $21.2 million for the nine months ended September 30, 2022. The increase resulted primarily from an increase of $14.4 million in sales to Chiesi, following the approvals by the FDA and the EMA of Elfabrio, which was partially offset by a decrease of $3.3 million in sales to Pfizer of $2.0 million in sales to Brazil, resulting from timing differences.
Revenues from License and R&D Services
We recorded revenues from license and R&D services of $24.7 million for the nine months ended September 30, 2023, an increase of $6.9 million, or 39%, compared to revenues of $17.8 million for the nine months ended September 30, 2022. The increase resulted from the $20.0 million regulatory milestone payment from Chiesi in connection with the FDA approval of Elfabrio which was partially offset by a decrease of $13.1 million in revenues recognized in connection with the Chiesi Agreements. Revenues from license and R&D services are comprised primarily of revenues we recognized in connection with the Chiesi Agreements.
Cost of Goods Sold
Cost of goods sold was $14.1 million for the nine months ended September 30, 2023, a decrease of $3.1 million, or 18%, from cost of goods sold of $17.2 million for the nine months ended September 30, 2022. The decrease in cost of goods sold was primarily the result of a decrease in sales of goods to Brazil and to Pfizer which was partially offset by an increase in sales of Elfabrio to Chiesi. Sales to Chiesi included certain drug substance costs which had already been recognized as research and development expenses as it was produced as part of research and development activities. Accordingly, the related cost of goods sold does not include the costs of such drug substance.
Research and Development Expenses
For the nine months ended September 30, 2023, our total research and development expenses were approximately $14.0 million comprised of approximately $6.2 million in subcontractor-related expenses, approximately $5.5 million of salary and related expenses, approximately $0.5 million of materials-related expenses and approximately $1.8 million of other expenses. For the nine months ended September 30, 2022, our total research and development expenses were approximately $23.7 million comprised of approximately $15.1 million in subcontractor-related expenses, approximately $5.4 million of salary and related expenses, approximately $1.2 million of materials-related expenses and approximately $2.0 million of other expenses.
Total decrease in research and developments expenses was $9.7 million, or 41%, for the nine months ended September 30, 2023 compared to the nine months ended September 30, 2022. The decrease in research and development expenses primarily resulted from the completion of our Fabry clinical program and of the regulatory processes related to the BLA and MAA review of Elfabrio by the applicable regulatory agencies.
We expect research and development expenses to continue to be our primary expense as we enter into a more advanced stage of preclinical and clinical trials for certain of our product candidates.
Selling, General and Administrative Expenses
Selling, general and administrative expenses were $10.8 million for the nine months ended September 30, 2023, an increase of $2.2 million, or 26%, compared to $8.6 million for the nine months ended September 30, 2022. The increase resulted primarily from an increase of approximately $1.5 million in salary and related expenses due to one-time cash bonuses and increase in share-based compensation, an increase of $0.2 million in professional fees and of $0.3 million in travel, conferences and employee training expenses.
Financial Expenses, Net
Financial expenses, net were $1.1 million for the nine months ended September 30, 2023, an increase of $0.4 million, or 57%, compared to $0.7 million for the nine months ended September 30, 2022. The increase was primarily due to a decrease of $0.6 million
31
in income related to exchange rates as well as an increase in our convertible notes related expenses of $0.5 million net of a gain recognized due to conversions of a portion of the 2024 Notes of $0.4 million and a $0.3 million increase in interest income.
Income taxes
In the nine months ended September 30, 2023, we recorded income taxes of approximately $0.6 million which were primarily the result of the provision for current taxes in respect of Section 174 of the TCJA. Section 174 eliminated the option to immediately deduct research and development expenses in the year incurred and requires us to capitalize and amortize these expenditures over 15 years (for out of U.S.-based research and development). In addition, during the nine months ended September 30, 2023, we released a valuation allowance related to deferred tax assets of the U.S. jurisdiction that resulted in a net benefit to tax expense of $3.1 million.
Liquidity and Capital Resources
Our sources of liquidity includes our cash and cash equivalents balance. At September 30, 2023, we had $41.0 million in cash, cash equivalents and short term bank deposits. We have primarily financed our operations through equity and debt financings, business collaborations, and grants funding.
During the year ended December 31, 2022, we raised gross proceeds equal to approximately $8.7 million from the sale of 7,473,038 shares of our common stock under our ATM program. During the six months ended June 30, 2023, we raised gross proceeds equal to approximately $24.9 million from sales of common stock under our ATM program through the sale of 12,560,150 shares of our common stock of which 4,347,668 shares were sold; we did not sell any common stock under our ATM program during the three-months ended September 30, 2023.
On August 25, 2021, we completed exchanges, or the Exchanges, of a substantial majority of our then outstanding 7.50% Senior Secured Convertible Notes due 2021, or the 2021 Notes, with institutional note holders of a substantial majority of the 2021 Notes. The Exchanges involved the exchange of an aggregate of $54.65 million principal amount of our 2021 Notes for an aggregate of $28.75 million principal amount of newly issued 7.50% Senior Secured Convertible Notes due 2024, or the 2024 Notes, $25.90 million in cash, and approximately $1.1 million in cash representing accrued and unpaid interest through the closing date. The initial conversion rate for the 2024 Notes is 563.2216 shares of our common stock for each $1,000 principal amount of 2024 Notes (equivalent to an initial conversion price of approximately $1.7755 per share of common Stock, subject to adjustment in certain circumstances. This initial conversion price represents a premium of approximately 32.5% relative to the closing price of the common stock on the NYSE American on August 13, 2021. After giving effect to the Exchanges, $3.27 million aggregate principal amount of the 2021 Notes remained outstanding. On November 15, 2021, all of the then outstanding 2021 Notes matured and were paid in full. As a result of the conversion of 2024 Notes during the nine months ended September 30, 2023, the total principal amount of our remaining 2024 Notes outstanding decreased by approximately $8.3 million. As of September 30, 2023, the total principal amount of our 2024 Notes outstanding was $20.42 million.
The 2024 Notes were issued pursuant to the Indenture dated as of August 24, 2021 between us, the guarantors party thereto, The Bank of New York Mellon Trust Company, N.A., as trustee and Wilmington Savings Fund Society, FSB, as collateral agent, or the 2024 Indenture. Interest on the Notes are payable semi-annually at a rate of 7.50% per annum. The 2024 Notes will mature on September 1, 2024, unless earlier purchased, converted, exchanged or redeemed, and are guaranteed by our subsidiaries. The 2024 Notes are secured by perfected liens on all of our assets, including those of our subsidiaries. Under the terms of the 2024 Indenture, we are required to comply with certain covenants, including the requirement to maintain a minimum cash balance of at least $7.5 million. Failure to comply with such covenants may result in an event of default under the 2024 Indenture and, accordingly, may result in the acceleration of the payment of the notes or in additional interest payments. As of September 30, 2023, we were in compliance with all covenants.
We believe that our cash, cash equivalents and short term bank deposits as of September 30, 2023 are sufficient to satisfy our capital needs for at least 12 months from the date that these financial statements are issued.
Cash Flows
Net cash used in operations was $4.9 million for the nine months ended September 30, 2023. The net income for the nine months ended September 30, 2023 of $14.4 million was decreased by a $13.2 million decrease in contracts liability, a $3.1 million increase in deferred tax assets, a $4.8 million increase in inventories, a $4.2 million increase in accounts receivable-trade and other assets and a $0.5 million financial income-net (mainly currency exchange differences). The net income was increased by a $3.8 million increase in accounts payable and accruals, $2.1 million in share-based compensation and $0.9 million in depreciation. Net cash used in investing activities for the nine months ended September 30, 2023 was $16.4 million and consisted primarily of net investment in bank deposits.
32
Net cash provided by financing activities was $24.7 million resulting primarily from the sale of common stock under our ATM program.
Net cash used in operations was $22.4 million for the nine months ended September 30, 2022. The net loss for the nine months ended September 30, 2022 of $11.2 million was increased by a $5.5 million decrease in contracts liability, a $4.4 million decrease in accounts payable and accruals, a $5.7 million increase in accounts receivable and other assets and an increase of $1.1 million in financial income, net (mainly exchange differences), and was partially offset by a $3.4 million decrease in inventories and a $1.5 million in share-based compensation. Net cash used in investing activities for the nine months ended September 30, 2022 was $10.0 million and consisted primarily of net investment in bank deposits. Net cash provided by financing activities was $4.2 million resulting mainly from the sale of common stock under our ATM program.
As a result of our significant research and development expenditures and the lack of significant revenue from sales of taliglucerase alfa, we have generated operating losses from our continuing operations since our inception. Our outstanding 2024 Notes are secured by a perfected lien on all of our assets. Under the terms of the 2024 Indenture, we are required to comply with certain covenants, including the requirement to maintain a minimum cash balance of at least $7.5 million. Failure to comply with such covenants may result in an event of default under the 2024 Indenture and, accordingly, may result in the acceleration of the payment of the notes or in additional interest payments. As of September 30, 2023, we were in compliance with all covenants.
During the nine months ended September 30, 2023, we issued, in the aggregate, 538,822 shares of our common stock in connection with cash and cashless exercises of our outstanding warrants to purchase shares of our common stock at an exercise price equal to $2.36 per share which were initially issued on March 18, 2020 as part of our private placement of common stock and warrants, or the 2020 Warrants. In connection with the exercises, we generated net proceeds equal to $0.7 million and retired 2020 Warrants to purchase 1,146,810 shares of common stock. As of September 30, 2023, 2020 Warrants to purchase 13,439,712 shares were outstanding.
In addition, during the nine months ended September 30, 2023, we issued, in the aggregate, 4,691,623 shares of our common stock in connection with a number of conversions of 2024 Notes. In connection with such conversions, during the nine months ended September 30, 2023, we paid to the converting holders $0.9 million representing cash payments due to accrued but unpaid interest, make-whole interest payments and payments in lieu of fractional shares. As a result of the conversion of 2024 Notes during the nine months ended September 30, 2023, the total principal amount of our remaining 2024 Notes outstanding decreased by approximately $8.3 million. As of September 30, 2023, the total principal amount of our 2024 Notes outstanding was $20.42 million.
Future Funding Requirements
We expect to continue to incur expenditures in the near future as we increase our research and developments efforts with respect to our product candidates. We cannot anticipate the costs or the timing of the occurrence of such costs. We also expect to increase the revenues generated from the sales of our approved drug products. To the extent we need to obtain additional financing in excess of such anticipated revenues, it may be more difficult for us to do so given the volatility of the price of our common stock. Our material cash needs for the next 24 months will include, among other expenses, (i) costs of preclinical and clinical trials, (ii) employee salaries, (iii) payments for rent and operation of our manufacturing facilities, (iv) fees to our consultants and legal advisors, patent advisors and fees for service providers in connection with our research and development efforts, (v) payments of principal and interest on our outstanding 2024 Notes and (vi) tax payments. We believe that the funds currently available to us are sufficient to satisfy our capital needs for at least 12 months.
As discussed above, we may be required to raise additional capital to develop our product candidates and continue research and development activities. Our ability to raise capital, and the amounts of necessary capital, will depend on many other factors, including:
| ● | the duration and cost of discovery and preclinical development and laboratory testing and clinical trials for our product candidates; |
| ● | Chiesi’s progress in commercializing Elfabrio; |
| ● | our progress in commercializing BioManguinhos alfataliglicerase in Brazil; |
| ● | the timing and outcome of regulatory review of our product candidates; |
| ● | the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing patent claims and other intellectual property rights; and |
33
| ● | the costs associated with any litigation claims. |
We expect to finance our future cash needs through sales of our approved drug products (Elelyso and Elfabrio), corporate collaborations, licensing or similar arrangements, public or private equity offerings and/or debt financings. We currently do not have any commitments for future external funding, except with respect to milestone payments and royalties that may become payable under the Chiesi Agreements. As of September 30, 2023, shares of our common stock for total gross proceeds of approximately $6.4 million remain available to be sold under our 2023 Sales Agreement.
Effects of Currency Fluctuations
Currency fluctuations could affect us through increased or decreased acquisition costs for certain goods and services and salaries expenses. We do not believe currency fluctuations have had a material effect on our results of operations during the three and nine months ended September 30, 2023.
Off-Balance Sheet Arrangements
We have no off-balance sheet arrangements as of each of September 30, 2023 and December 31, 2022.
Item 3. Quantitative and Qualitative Disclosures about Market Risk
Currency Exchange Risk
The currency of the primary economic environment in which our operations are conducted is the U.S. dollar. Most of our revenues and more than 50% of our expenses and capital expenditures are and were incurred in dollars, and a significant source of our financing has been provided in U.S. dollars. Since the dollar is the functional currency, monetary items maintained in currencies other than the dollar are remeasured using the rate of exchange in effect at the balance sheet dates and non-monetary items are remeasured at historical exchange rates. Revenue and expense items are remeasured at the average rate of exchange in effect during the period in which they occur. Foreign currency translation gains or losses are recognized in the statement of operations.
Approximately 43% of our costs, including salaries, expenses and office expenses, are incurred in NIS. Inflation in Israel may have the effect of increasing the U.S. dollar cost of our operations in Israel. If the U.S. dollar declines in value in relation to the NIS, it will become more expensive for us to fund our operations in Israel. A revaluation of 1% of the NIS will affect our loss before tax by less than 1%. The exchange rate of the U.S. dollar to the NIS, based on exchange rates published by the Bank of Israel, was as follows:
Three Months Ended | Nine Months Ended | |||||||||
September 30, | September 30, | December 31, | ||||||||
2023 |
| 2022 | 2023 |
| 2022 |
| 2022 | |||
Average rate for period | 3.745 |
| 3.402 | 3.643 |
| 3.315 |
| 3.360 | ||
Rate at period-end | 3.824 |
| 3.543 | 3.824 |
| 3.543 |
| 3.519 | ||
To date, we have not engaged in hedging transactions. In the future, we may enter into currency hedging transactions to decrease the risk of financial exposure from fluctuations in the exchange rate of the U.S. dollar against the NIS. These measures, however, may not adequately protect us from material adverse effects due to the impact of inflation in Israel.
Item 4. Controls and Procedures
Evaluation of Disclosure Controls and Procedures
We conducted an evaluation of the effectiveness of the design and operation of our disclosure controls and procedures as of the end of the period covered by this Quarterly Report on Form 10-Q. The evaluation was conducted under the supervision and with the participation of management, including our Chief Executive Officer and Chief Financial Officer. Disclosure controls and procedures are controls and procedures designed to reasonably assure that information required to be disclosed in our reports filed under the Exchange Act, such as this Quarterly Report on Form 10-Q, is recorded, processed, summarized and reported within the time periods specified in the Commission’s rules and forms. Disclosure controls and procedures are also designed to reasonably assure that such information is accumulated and communicated to our management, including the Chief Executive Officer and Chief Financial Officer, as appropriate to allow timely decisions regarding required disclosure.
Based on the evaluation, our Chief Executive Officer and Chief Financial Officer have concluded that, as of the end of the period covered by this Quarterly Report on Form 10-Q, our disclosure controls and procedures were effective to provide reasonable
34
assurance that information required to be disclosed in our Exchange Act reports is recorded, processed, summarized and reported within the time periods specified by the Commission, and that material information relating to our company and our consolidated subsidiary is made known to management, including the Chief Executive Officer and Chief Financial Officer, particularly during the period when our periodic reports are being prepared.
Inherent Limitations on Effectiveness of Controls
Our management, including our Chief Executive Officer and Chief Financial Officer, does not expect that our disclosure controls and procedures or our internal control over financial reporting will prevent or detect all error and all fraud. A control system, no matter how well designed and operated, can provide only reasonable, not absolute, assurance that the control system’s objectives will be met. The design of a control system must reflect the fact that there are resource constraints, and the benefits of controls must be considered relative to their costs. Further, because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that misstatements due to error or fraud will not occur or that all control issues and instances of fraud, if any, within a company have been detected.
Changes in Internal Control over Financial Reporting
There were no changes in our internal control over financial reporting (as defined in Rules 13a-15f and 15d-15f under the Exchange Act) that occurred during the quarter ended September 30, 2023 that have materially affected, or that are reasonably likely to materially affect, our internal control over financial reporting.
35
PART II – OTHER INFORMATION
Item 1. Legal Proceedings
We are not involved in any material legal proceedings.
Item 1A. Risk Factors
Except as set forth below, there have been no material changes to the risk factors previously disclosed in our Annual Report on Form 10-K for the year ended December 31, 2022 and in our Quarterly Reports on Form 10-Q for the three-month periods ended March 31, 2023 and June 30, 2023.
Significant parts of our operations are located in Israel and, therefore, our results may be adversely affected by political, economic and military conditions in Israel.
Our executive office and operations are located in the State of Israel. Accordingly, economic, geopolitical and military conditions in Israel directly affect our business. Any armed conflicts, political instability, terrorism, cyberattacks or any other hostilities involving Israel or the interruption or curtailment of trade between Israel and its present trading partners could affect adversely our operations. In October 2023, terrorists from the Hamas organization infiltrated Israel’s southern border from the Gaza Strip and in other areas within the State of Israel attacking a number of civilian and military targets while simultaneously launching extensive rocket attacks on the Israeli population and industrial centers. At the same time, clashes between Israel and Hezbollah in Lebanon have increased. In response, Israel’s security cabinet declared war against the Hamas and a military campaign against these terrorist organizations commenced in parallel to their continued rocket and terror attacks. The attacks by Hamas and Hezbollah, and Israel’s defensive measures, may result in a greater regional conflict. To date, our operations have not been adversely affected by this situation. Hostilities have not taken place where our facilities are located and we do not anticipate any disruption to the supply of Elfabrio or Elelyso. However, our facilities are in range of rockets that were fired from Lebanon into Israel during a 2006 war with the Hezbollah in Lebanon, and suffered minimal damages during one of the rocket attacks. Our insurance policies do not cover damages incurred in connection with these conflicts or for any resulting disruption in our operations. The Israeli government, as a matter of law, provides coverage for the reinstatement value of direct damages that are caused by terrorist attacks or acts of war; however, the government may cease providing such coverage or the coverage might not be enough to cover potential damages. If our facilities are damaged as a result of hostile action, our operations may be materially adversely affected.
Ongoing and revived hostilities or other Israeli political or economic factors, could prevent or delay future shipments of our products, harm our operations and product development and cause any future sales to decrease. In the event that hostilities disrupt the ongoing operation of our facilities or the airports and seaports on which we depend to import and export our supplies, materials, drug substance and other products, our operations may be materially adverse affected. It is currently not possible to predict the duration or severity of the ongoing conflict or its effects on our business, operations and financial conditions. The ongoing conflict is rapidly evolving and developing, and could disrupt certain of our business and operations, among others
Our operations may be disrupted by the obligations of our personnel to perform military service which could have a material adverse effect on our business.
Many of our male employees in Israel, including members of senior management, are obligated to perform up to one month (in some cases more) of annual military reserve duty until they reach the age of 45 and, in the event of a military conflict, could be called to active duty. To date, we have not suffered any disruptions to our operations from the absence of employees in connection with the recent military action. However, we continue to face the risk that our operations could be disrupted by the absence of a significant number of our employees related to military service or the absence for extended periods of military service of one or more of our key employees. Any such disruption may have a material adverse effect on our business, results of operations and financial condition.
Item 2. Unregistered Sales of Equity Securities and Use of Proceeds
None.
Item 3. Defaults Upon Senior Securities
None.
36
Item 4. Mine Safety Disclosure
Not applicable.
Item 5. Other Information
During the quarter ended September 30, 2023, none of our directors or officers adopted or terminated a Rule 10b5-1 trading arrangement or non-Rule 10b5-1 trading arrangement (as such terms are defined in Item 408 of Regulation S-K).
.
Item 6. Exhibits
Incorporated by Reference | |||||||
Exhibit Number |
| Exhibit Description | Form | File Number | Exhibit | Date | Filed or Furnished Herewith |
3.1 | 8-K | 001-33357 | 3.1 | April 1, 2016 | |||
3.2 | Def 14A | 001-33357 | Appen. A | July 1, 2016 | |||
3.3 | Second Amendment to Certificate of Incorporation of the Company | Def 14A | 001-33357 | Appen. A | October 17, 2018 | ||
3.4 | Third Amendment to Certificate of Incorporation of the Company | 8-K | 001-33357 | 3.1 | December 19, 2019 | ||
3.5 | Fourth Amendment to Certificate of Incorporation of the Company | 10-Q | 001-33357 | 3.5 | August 15, 2022 | ||
3.6 | Fifth Amendment to Certificate of Incorporation of the Company | 10-Q | 001-33357 | 3.6 | August 7, 2023 | ||
3.7 | 8-K | 001-33357 | 3.2 | April 1, 2016 | |||
4.1† | 8-K | 001-33357 | 4.1 | July 18, 2012 | |||
4.2 | 10-K | 001-33357 | 4.7 | February 27, 2023 | |||
4.3 | 8-K | 001-33357 | 4.1 | March 12, 2020 | |||
4.4† | 10-Q | 001-33357 | 4.8 | August 10, 2020 | |||
4.5 | 10-Q | 001-33357 | 4.9 | August 10, 2020 | |||
4.6 | 8-K | 001-33357 | 4.2 | August 26, 2021 | |||
4.7 | 8-K | 001-33357 | 4.3 | August 26, 2021 | |||
10.1† | Amended and Restated Pro BioTherapeutics, Inc. 2006 Stock Incentive Plan, as amended | 10-Q | 001-33357 | 10.1 | August 7, 2023 | ||
31.1 | X | ||||||
31.2 | X | ||||||
32.1 | X | ||||||
32.2 | X | ||||||
37
101.INS | XBRL Instance Document - the instance document does not appear in the Interactive Data File because its XBRL tags are embedded within the Inline XBRL document | X | |||||
101.SCH | Inline XBRL Taxonomy Extension Schema Document | X | |||||
101.CAL | Inline XBRL Taxonomy Extension Calculation Linkbase Document | X | |||||
101.DEF | Inline XBRL Taxonomy Extension Definition Linkbase Document | X | |||||
101.LAB | Inline XBRL Taxonomy Extension Labels Linkbase Document | X | |||||
101.PRE | Inline XBRL Taxonomy Extension Presentation Linkbase Document | X | |||||
104 | COVER PAGE INTERACTIVE DATA FILE (formatted as Inline XBRL and contained in Exhibit 101). | ||||||
† Management contracts or compensation plans or arrangements in which directors or executive officers are eligible to participate. | |||||||
38
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, as amended, the registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
PROTALIX BIOTHERAPEUTICS, INC. | ||
(Registrant) | ||
Date: November 6, 2023 | By: | /s/ Dror Bashan |
Dror Bashan President and Chief Executive Officer (Principal Executive Officer) | ||
Date: November 6, 2023 | By: | /s/ Eyal Rubin |
Eyal Rubin Senior Vice President and Chief Financial Officer, Treasurer and Secretary (Principal Financial and Accounting Officer) | ||
39
EXHIBIT 31.1
CERTIFICATION
I, Dror Bashan, certify that:
1. I have reviewed this quarterly report on Form 10-Q of Protalix BioTherapeutics, Inc.;
2. Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report;
3. Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this report;
4. The registrant’s other certifying officer and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the registrant and have:
(a) Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared;
(b) Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles;
(c) Evaluated the effectiveness of the registrant’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and
(d) Disclosed in this report any change in the registrant’s internal control over financial reporting that occurred during the registrant’s most recent fiscal quarter (the registrant’s fourth fiscal quarter in the case of an annual report) that has materially affected, or is reasonably likely to materially affect, the registrant’s internal control over financial reporting; and
5. The registrant’s other certifying officer and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the registrant’s auditors and the audit committee of the registrant’s board of directors (or persons performing the equivalent functions):
a) All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the registrant’s ability to record, process, summarize and report financial information; and
b) Any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant’s internal control over financial reporting.
Date: November 6, 2023 | |
| |
/s/ Dror Bashan | |
Dror Bashan | |
President and Chief Executive Officer | |
EXHIBIT 31.2
CERTIFICATION
I, Eyal Rubin, certify that:
1. I have reviewed this quarterly report on Form 10-Q of Protalix BioTherapeutics, Inc.;
2. Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report;
3. Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this report;
4. The registrant’s other certifying officer and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the registrant and have:
(a) Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared;
(b) Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles;
(c) Evaluated the effectiveness of the registrant’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and
(d) Disclosed in this report any change in the registrant’s internal control over financial reporting that occurred during the registrant’s most recent fiscal quarter (the registrant’s fourth fiscal quarter in the case of an annual report) that has materially affected, or is reasonably likely to materially affect, the registrant’s internal control over financial reporting; and
5. The registrant’s other certifying officer and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the registrant’s auditors and the audit committee of the registrant’s board of directors (or persons performing the equivalent functions):
a) All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the registrant’s ability to record, process, summarize and report financial information; and
b) Any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant’s internal control over financial reporting.
Date: November 6, 2023 | |
| |
/s/ Eyal Rubin | |
Eyal Rubin | |
Sr. Vice President & Chief Financial Officer, | |
Treasurer | |
EXHIBIT 32.1
PROTALIX BIOTHERAPEUTICS, INC.
CERTIFICATION
In connection with the quarterly report of Protalix BioTherapeutics, Inc. (the “Company”) on Form 10-Q for the period ended September 30, 2023 as filed with the Securities and Exchange Commission (the “Report”), I, Dror Bashan, President and Chief Executive Officer of the Company, hereby certify as of the date hereof, solely for purposes of Title 18, Chapter 63, Section 1350 of the United States Code, that to the best of my knowledge:
(1) the Report fully complies with the requirements of Section 13(a) or 15(d), as applicable, of the Securities Exchange Act of 1934; and
(2) the information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of the Company at the dates and for the periods indicated.
This Certification has not been, and shall not be deemed, “filed” with the Securities and Exchange Commission.
Date: November 6, 2023 | |
| |
/s/ Dror Bashan | |
Dror Bashan | |
President and Chief Executive Officer | |
EXHIBIT 32.2
PROTALIX BIOTHERAPEUTICS, INC.
CERTIFICATION
In connection with the quarterly report of Protalix BioTherapeutics, Inc. (the “Company”) on Form 10-Q for the period ended September 30, 2023 as filed with the Securities and Exchange Commission (the “Report”), I, Eyal Rubin, Senior Vice President and Chief Financial Officer of the Company, hereby certify as of the date hereof, solely for the purposes of Title 18, Chapter 63, Section 1350 of the United States Code, that to the best of my knowledge:
(1) the Report fully complies with the requirements of Section 13(a) or 15(d), as applicable, of the Securities Exchange Act of 1934; and
(2) the information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of the Company at the dates and for the periods indicated.
This Certification has not been, and shall not be deemed, “filed” with the Securities and Exchange Commission.
Date: November 6, 2023 | |
| |
/s/ Eyal Rubin | |
Eyal Rubin | |
Senior Vice President and Chief Financial Officer | |
CONDENSED CONSOLIDATED STATEMENTS OF OPERATIONS - USD ($) $ in Thousands |
3 Months Ended | 9 Months Ended | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
Sep. 30, 2023 |
Sep. 30, 2022 |
Sep. 30, 2023 |
Sep. 30, 2022 |
|||||||
| TOTAL REVENUE | $ 10,345 | $ 14,183 | $ 55,008 | $ 39,021 | ||||||
| COST OF GOODS SOLD | [1] | (4,893) | (7,074) | (14,126) | (17,195) | |||||
| RESEARCH AND DEVELOPMENT EXPENSES | [2] | (3,669) | (7,386) | (13,991) | (23,732) | |||||
| SELLING, GENERAL AND ADMINISTRATIVE EXPENSES | [3] | (3,670) | (2,848) | (10,816) | (8,613) | |||||
| OPERATING INCOME (LOSS) | (1,887) | (3,125) | 16,075 | (10,519) | ||||||
| FINANCIAL EXPENSES | (460) | (639) | (2,406) | (1,879) | ||||||
| FINANCIAL INCOME | 628 | 197 | 1,323 | 1,211 | ||||||
| FINANCIAL INCOME (EXPENSES), NET | 168 | (442) | (1,083) | (668) | ||||||
| INCOME (LOSS) BEFORE TAXES ON INCOME | (1,719) | (3,567) | 14,992 | (11,187) | ||||||
| TAXES ON INCOME | (133) | (636) | ||||||||
| NET INCOME (LOSS) FOR THE PERIOD | $ (1,852) | $ (3,567) | $ 14,356 | $ (11,187) | ||||||
| EARNINGS (LOSS) PER SHARE OF COMMON STOCK - BASIC | $ (0.03) | $ (0.07) | $ 0.22 | $ (0.24) | ||||||
| EARNINGS (LOSS) PER SHARE OF COMMON STOCK - DILUTED | $ (0.04) | $ (0.07) | $ 0.16 | $ (0.24) | ||||||
| WEIGHTED AVERAGE NUMBER OF SHARES OF COMMON STOCK USED IN COMPUTING EARNINGS (LOSS) PER SHARE- BASIC | 72,281,681 | 49,498,105 | 65,811,506 | 47,582,733 | ||||||
| WEIGHTED AVERAGE NUMBER OF SHARES OF COMMON STOCK USED IN COMPUTING EARNINGS (LOSS) PER SHARE- DILUTED | 83,782,679 | 49,498,105 | 81,040,281 | 47,582,733 | ||||||
| Goods | ||||||||||
| TOTAL REVENUE | $ 10,168 | $ 8,812 | $ 30,309 | $ 21,222 | ||||||
| License and R&D Services | ||||||||||
| TOTAL REVENUE | $ 177 | $ 5,371 | $ 24,699 | $ 17,799 | ||||||
| ||||||||||
CONDENSED CONSOLIDATED STATEMENTS OF OPERATIONS (Parenthetical) - USD ($) $ in Thousands |
3 Months Ended | 9 Months Ended | ||
|---|---|---|---|---|
Sep. 30, 2023 |
Sep. 30, 2022 |
Sep. 30, 2023 |
Sep. 30, 2022 |
|
| Cost of goods sold | ||||
| Share-based Compensation Arrangement by Share-based Payment Award, Compensation Cost [Line Items] | ||||
| Share-based compensation | $ 195 | $ 36 | $ 299 | $ 58 |
| Research and development expenses | ||||
| Share-based Compensation Arrangement by Share-based Payment Award, Compensation Cost [Line Items] | ||||
| Share-based compensation | 182 | 114 | 506 | 275 |
| Selling, general and administrative expenses | ||||
| Share-based Compensation Arrangement by Share-based Payment Award, Compensation Cost [Line Items] | ||||
| Share-based compensation | $ 720 | $ 272 | $ 1,276 | $ 1,213 |
CONDENSED CONSOLIDATED STATEMENTS OF CHANGES IN STOCKHOLDERS' EQUITY (CAPITAL DEFICIENCY) (Parenthetical) - $ / shares |
Sep. 30, 2023 |
Jul. 25, 2023 |
Jul. 24, 2023 |
Dec. 31, 2022 |
|---|---|---|---|---|
| CONDENSED CONSOLIDATED STATEMENTS OF CHANGES IN STOCKHOLDERS EQUITY (CAPITAL DEFICIENCY) | ||||
| Common Stock, Par or Stated Value Per Share | $ 0.001 | $ 0.001 | ||
| Common Stock, Shares Authorized | 185,000,000 | 185,000,000 | 144,000,000 | 144,000,000 |
CONDENSED CONSOLIDATED STATEMENTS OF CASH FLOWS - USD ($) $ in Thousands |
9 Months Ended | |
|---|---|---|
Sep. 30, 2023 |
Sep. 30, 2022 |
|
| CASH FLOWS FROM OPERATING ACTIVITIES: | ||
| Net income (loss) | $ 14,356 | $ (11,187) |
| Adjustments required to reconcile net income (loss) to net cash used in operating activities: | ||
| Share-based compensation | 2,081 | 1,546 |
| Depreciation | 878 | 811 |
| Financial income, net (mainly exchange differences) | (511) | (1,083) |
| Changes in accrued liability for employee rights upon retirement | (50) | (391) |
| Changes in deferred tax asset | (3,092) | |
| Loss (gain) on amounts funded in respect of employee rights upon retirement | (49) | 6 |
| Gain on conversions of convertible notes | (421) | |
| Amortization of debt issuance costs and debt discount | 208 | 224 |
| Changes in operating assets and liabilities: | ||
| Decrease in contracts liability | (13,178) | (5,547) |
| Increase in accounts receivable-trade and other assets | (4,188) | (5,692) |
| Changes in operating lease right of use assets, net | 12 | (30) |
| Decrease (increase) in inventories | (4,779) | 3,392 |
| Increase (decrease) in accounts payable and accruals | 3,820 | (4,438) |
| Net cash used in operating activities | (4,913) | (22,389) |
| CASH FLOWS FROM INVESTING ACTIVITIES: | ||
| Investment in bank deposits | (20,420) | (16,000) |
| Proceeds from sale of short-term deposits | 5,000 | 6,000 |
| Purchase of property and equipment | (899) | (415) |
| Amounts paid (funded) in respect of employee rights upon retirement, net | (50) | 427 |
| Net cash used in investing activities | (16,369) | (9,988) |
| CASH FLOWS FROM FINANCING ACTIVITIES: | ||
| Proceeds from issuance of common stock under the Sales Agreement, net | 23,954 | 4,161 |
| Exercise of warrants | 712 | 2 |
| Net cash provided by financing activities | 24,666 | 4,163 |
| EFFECT OF EXCHANGE RATE CHANGES ON CASH AND CASH EQUIVALENTS | (87) | (51) |
| NET INCREASE (DECREASE) IN CASH AND CASH EQUIVALENTS | 3,297 | (28,265) |
| BALANCE OF CASH AND CASH EQUIVALENTS AT BEGINNING OF PERIOD | 17,111 | 38,985 |
| BALANCE OF CASH AND CASH EQUIVALENTS AT END OF PERIOD | 20,408 | 10,720 |
| SUPPLEMENTARY INFORMATION ON INVESTING AND FINANCING ACTIVITIES NOT INVOLVING CASH FLOWS: | ||
| Purchase of property and equipment | 253 | 205 |
| Operating lease right of use assets obtained in exchange for new operating lease liabilities | 1,395 | 396 |
| Convertible notes conversions | 7,783 | |
| SUPPLEMENTARY DISCLOSURE ON CASH FLOWS | ||
| Interest paid | 2,743 | 2,198 |
| Interest received | $ 303 | $ 136 |
SIGNIFICANT ACCOUNTING POLICIES |
9 Months Ended | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Sep. 30, 2023 | ||||||||||||||||
| SIGNIFICANT ACCOUNTING POLICIES | ||||||||||||||||
| SIGNIFICANT ACCOUNTING POLICIES | NOTE 1 - SIGNIFICANT ACCOUNTING POLICIES
Protalix BioTherapeutics, Inc. (collectively with its subsidiaries, the “Company”) and its wholly-owned subsidiaries, Protalix Ltd. and Protalix B.V. (collectively, the “Subsidiaries”), are biopharmaceutical companies focused on the development and commercialization of recombinant therapeutic proteins based on the Company’s proprietary ProCellEx® protein expression system (“ProCellEx”). The Company’s current focus is to facilitate the commercialization efforts of Chiesi Farmaceutici S.p.A. (“Chiesi”), the Company’s development and commercialization partner for pegunigalsidase alfa, or Elfabrio® (which the Company referred to as PRX-102 during its development stage), for the treatment of Fabry disease, a rare, genetic lysosomal disorder. To date, the Company has successfully developed taliglucerase alfa (marketed under the name Elelyso® except in Brazil where it is marketed as BioManguinhos alfataliglicerase), an enzyme replacement therapy for the treatment of Gaucher disease that has been approved for marketing in the United States, Brazil, Israel and other markets. The Company’s strategy is to develop proprietary recombinant proteins designed to address high, unmet needs in the rare disease space that are therapeutically superior to existing recombinant proteins currently marketed for the same indications. Consistent with this strategy, the Company has a number of product candidates in varying stages of the clinical development process. On May 5, 2023, the European Commission (“EC”) announced that it had approved the Marketing Authorization Application (“MAA”) for Elfabrio and on May 9, 2023, the U.S. Food and Drug Administration (“FDA”) announced that it had approved the Biologics License Application (“BLA”) for Elfabrio, each for adult patients with Fabry disease. Both approvals cover the 1 mg/kg every two weeks dosage. The European Medicines Agency (“EMA”) approval followed the February 2023 adoption of a positive opinion and recommendation of marketing authorization for Elfabrio by the EMA’s Committee for Medicinal Products for Human Use the (“CHMP”). Elfabrio was approved by the FDA with a boxed warning for hypersensitivity reactions/anaphylaxis, consistent with Enzyme Replacement Therapy (ERT) class labeling, and Warnings/Precautions providing guidance on the signs and symptoms of hypersensitivity and infusion-associated reactions seen in the clinical studies as well as treatments to manage such events should they occur. The Warnings/Precautions for membranoproliferative glomerulonephritis (MPGN) alert prescribers to the possibility of MPGN and provide guidance for appropriate patient management. Overall, the FDA review team concluded that in the context of Fabry disease as a rare, serious disease with limited therapeutic options that may not be suitable to all individual patients, the benefit-risk of Elfabrio is favorable for the treatment of adults with confirmed Fabry disease. In August and September of 2023, Elfabrio was approved in Great Britain and Switzerland, respectively, for long-term enzyme replacement therapy in adult patients with a confirmed diagnosis of Fabry disease. The Company has entered into two exclusive global licensing and supply agreements for Elfabrio with its development and commercialization partner for PRX-102, Chiesi Farmaceutici S.p.A. (“Chiesi”). On October 19, 2017, Protalix Ltd., the Company’s wholly-owned subsidiary, entered into an Exclusive License and Supply Agreement with Chiesi (the “Chiesi Ex-US Agreement”), pursuant to which Chiesi was granted an exclusive license for all markets outside of the United States to commercialize Elfabrio. On July 23, 2018, Protalix Ltd. entered into an Exclusive License and Supply Agreement with Chiesi (the Chiesi US Agreement, with respect to the commercialization of Elfabrio in the United States. Elfabrio, an enzyme replacement therapy, or ERT, was the subject of a phase III clinical program studying the drug as a treatment of patients with Fabry disease, a rare, genetic lysosomal disorder. The phase III clinical program included three separate studies, which are referred to as the BALANCE study, the BRIDGE study and the BRIGHT study. The phase III clinical program analyzed two potential dosing regimens: 1 mg/kg every two weeks and 2 mg/kg every four weeks. In addition, the phase III clinical program included two extension studies in which subjects that participated in our phase I/II clinical trials and phase III clinical trials had the opportunity to enroll and continue to be treated with PRX-102. As of March 1, 2023, sponsorship of the two open-label extension studies was transferred to Chiesi, which is now administering the extension studies. Over time, and as Elfabrio is approved for marketing in different jurisdictions, participants withdraw from the open-label extension studies. Some of the withdrawals transfer to a commercial setting, others withdraw for other reasons. The BLA for Elfabrio for the treatment of adult patients with Fabry disease was resubmitted to the FDA on November 9, 2022. An initial BLA for Elfabrio was submitted to the FDA on May 27, 2020 under the FDA’s Accelerated Approval pathway, but resulted in a Complete Response Letter (“CRL”). The MAA was submitted to the EMA on February 7, 2022, after the October 8, 2021 meeting we held, together with Chiesi, with the Rapporteur and Co-Rapporteur of the EMA regarding PRX-102. The FDA publicly released the internal review documents for Elfabrio (pegunigalsidase alfa-iwxj) injection BLA 761161. These documents provide previously unavailable additional information regarding the basis for the FDA’s May 2023 approval decision. In particular, the FDA determined that substantial evidence of effectiveness for Elfabrio in Fabry patients was established with one adequate and well-controlled study (Study PB-102-F01/02) with confirmatory evidence provided by the BALANCE study (also referred to as Study PB-102-F20). The FDA review team also concluded that the BALANCE study met its primary efficacy endpoint, which assessed the annualized rate of change in eGFR (estimated glomerular filtration rate) over 104 weeks. However, the FDA also determined that the results from the BALANCE study did not support a non-inferiority claim to the comparator product due to the lack of data to support a non-inferiority margin. The Company has licensed the rights to commercialize taliglucerase alfa worldwide (other than Brazil) to Pfizer Inc. (“Pfizer”), and in Brazil to Fundação Oswaldo Cruz (“Fiocruz”), an arm of the Brazilian Ministry of Health (the “Brazilian MoH”). Otherwise, except with respect to taliglucerase alfa and Elfabrio, the Company holds the worldwide commercialization rights to its other proprietary development candidates. In addition, the Company continuously evaluates potential strategic marketing partnerships as well as collaboration programs with biotechnology and pharmaceutical companies and academic research institutions. The Company’s product pipeline currently includes, among other candidates: (1)PRX-115, the Company’s plant cell-expressed recombinant PEGylated uricase (urate oxidase) – a chemically modified enzyme to treat severe gout; and (2)PRX-119, the Company’s plant cell-expressed PEGylated recombinant human DNase I product candidate being designed to elongate half-life in the circulation for NETs-related diseases. On March 21, 2023, the first patient was dosed in the Company’s phase I First-in-Human (“FIH”) clinical trial of PRX-115. As of September 30, 2023, 32 patients have been dosed in this trial. Obtaining marketing approval with respect to any product candidate in any country is dependent on the Company’s ability to implement the necessary regulatory steps required to obtain such approvals, and demonstrate the safety and efficacy of its product candidates. The Company cannot reasonably predict the outcome of these activities. On July 2, 2021, the Company entered into an At The Market Offering Agreement (the “2021 Sales Agreement”) with H.C. Wainwright & Co., LLC, as the Company’s sales agent (the “Agent”) which was amended on May 2, 2022. Pursuant to the terms of the 2021 Sales Agreement, the Company was able to sell, from time to time through the Agent, shares of its common stock, par value $0.001 per share (the “Common Stock”), having an aggregate offering price of up to $20.0 million (the “ATM Shares”). Upon execution of the 2021 Sales Agreement, the Company terminated the ATM Equity OfferingSM Sales Agreement it had entered into on October 1, 2020 with BofA Securities, Inc. (“BofA Securities”). During the term of the sales agreement with BofA Securities, the Company sold a total of 3,296,123 shares of Common Stock for total gross proceeds of approximately $13.8 million. During the term of the 2021 Sales Agreement which ended during the quarter ended March 31, 2023, the Company sold a total of 13,980,060 ATM Shares for total gross proceeds of approximately $20.0 million under the 2021 Sales Agreement, thereby completing the ATM program under said agreement. On February 27, 2023, the Company entered into an At The Market Offering Agreement (the “2023 Sales Agreement”) with the Agent. Pursuant to the terms of the 2023 Sales Agreement, the Company may sell, from time to time through the Agent, ATM Shares having an aggregate offering price of up to $20.0 million. As of September 30, 2023, shares of Common Stock for total gross proceeds of approximately $6.4 million remain available to be sold under the 2023 Sales Agreement. Under each of the Chiesi Ex-US Agreement and the Chiesi US Agreement (collectively, the “Chiesi Agreements”), Chiesi made an upfront payment to Protalix Ltd. of $25.0 million in connection with the execution of each agreement. In addition, under the Chiesi Ex-US Agreement, Protalix Ltd. is entitled to additional payments of up to $25.0 million in pegunigalsidase alfa development costs and to receive additional payments of up to $320.0 million, in the aggregate, in regulatory and commercial milestone payments. Under the Chiesi US Agreement, Protalix Ltd. is entitled to payments of up to a maximum of $20.0 million to cover development costs for pegunigalsidase alfa and to receive additional payments of up to a maximum of $760.0 million, in the aggregate, in regulatory and commercial milestone payments. To date, Protalix Ltd. has received the complete amount of development costs to which it is entitled under the Chiesi Agreements. In addition, following the approval of Elfabrio by the FDA, the Company received a milestone payment equal to $20.0 million. Under the terms of both of the Chiesi Agreements, Protalix Ltd. is required to manufacture all of the Elfabrio drug substance needed under the agreements, subject to certain exceptions, and Chiesi will purchase Elfabrio drug product from Protalix, subject to certain terms and conditions. The consideration for Protalix Ltd. is based on the drug product supplied to Chiesi and the average selling price of the drug product in the relevant territory multiplied by tiered payments as described in the relevant agreement. Under the Chiesi Ex-US Agreement, Chiesi is required to make tiered payments for drug product purchased from Protalix of 15% to 35%, depending on the amount of annual net sales outside of the United States, and under the Chiesi US Agreement, Chiesi is required to make tiered payments for drug product purchased from Protalix of 15% to 40%, depending on the amount of annual net sales in the United States. On August 29, 2022, the Company entered into a Fill/Finish Agreement (the “F/F Agreement”) and a Letter Agreement (the “Letter Agreement”), in each case with Chiesi. The Company agreed to supply Chiesi with drug substance for PRX-102 and, following relevant technology and technical information transfer activities, Chiesi has agreed, among other things, to provide the Company with commercial fill/finish services for PRX-102, including to support the anticipated global launch of PRX-102. The F/F Agreement shall continue in force until December 31, 2025, unless terminated earlier in accordance with the terms of the F/F Agreement and the term may be extended by mutual agreement for an additional period of seven years upon mutual written agreement prior to expiration of the initial term. On May 13, 2021, the Company signed a binding term sheet with Chiesi pursuant to which the Company and Chiesi amended the Chiesi Agreements in order to provide the Company with near-term capital. Chiesi agreed to make a $10.0 million payment to the Company before the end of the second quarter of 2021 in exchange for a $25.0 million reduction in a longer term regulatory milestone payment provided in the Chiesi EX-US Agreement. All other regulatory and commercial milestone payments remain unchanged. The Company received the payment in June 2021. The Company also agreed to negotiate certain manufacturing related matters. Since its approval by the FDA, taliglucerase alfa has been marketed by Pfizer in accordance with the exclusive license and supply agreement entered into between Protalix Ltd. and Pfizer, which is referred to herein as the Pfizer Agreement. In October 2015, Protalix Ltd. and Pfizer entered into an amended exclusive license and supply agreement, which is referred to herein as the Amended Pfizer Agreement, pursuant to which the Company sold to Pfizer its share in the collaboration created under the Pfizer Agreement for the commercialization of Elelyso. As part of the sale, the Company agreed to transfer its rights to Elelyso in Israel to Pfizer while gaining full rights to it in Brazil. Under the Amended Pfizer Agreement, Pfizer is entitled to all of the revenues, and is responsible for 100% of expenses globally for Elelyso, excluding Brazil where the Company is responsible for all expenses and retains all revenues. On June 18, 2013, the Company entered into a Supply and Technology Transfer Agreement with Fiocruz (the “Brazil Agreement”) for taliglucerase alfa. Fiocruz’s purchases of BioManguinhos alfataliglicerase to date have been significantly below certain agreed-upon purchase milestones and, accordingly, the Company has the right to terminate the Brazil Agreement. Notwithstanding the termination right, the Company is, at this time, continuing to supply BioManguinhos alfataliglicerase to Fiocruz and patients continue to be treated with BioManguinhos alfataliglicerase in Brazil. The Company expects to continue to incur significant expenditures in the near future due to research and developments efforts with respect to the product candidates. Under the terms of the Company’s outstanding 7.50% Senior Secured Convertible Notes due 2024 (the “2024 Notes”), the Company is required to comply with certain financial covenants, including the maintenance of a minimum cash balance of at least $7.5 million. As of September 30, 2023, the Company is in compliance with all such covenants. The Company believes that its cash and cash equivalents as of September 30, 2023, are sufficient to satisfy the Company’s capital needs for at least 12 months from the date that these financial statements are issued.
The accompanying unaudited condensed consolidated financial statements of the Company have been prepared in accordance with generally accepted accounting principles in the United States (“GAAP”) for interim financial information. Accordingly, they do not include all of the information and notes required by GAAP for annual financial statements. In the opinion of management, all adjustments (of a normal recurring nature) considered necessary for a fair statement of the results for the interim periods presented have been included. Operating results for the interim period are not necessarily indicative of the results that may be expected for the full year. These unaudited condensed consolidated financial statements should be read in conjunction with the audited consolidated financial statements in the Annual Report on Form 10-K for the year ended December 31, 2022, filed by the Company with the U.S. Securities and Exchange Commission (the “Commission”). The comparative balance sheet at December 31, 2022 has been derived from the audited financial statements at that date. There have been no material changes in our significant accounting policies as described in our consolidated financial statements for the year ended December 31, 2022.
The Company accounts for revenue pursuant to Accounting Standards Codification, Topic 606, Revenue from Contracts with Customers (“ASC 606”). Under ASC 606, a contract with a customer exists only when: the parties to the contract have approved it and are committed to perform their respective obligations, the Company can identify each party’s rights regarding the distinct goods or services to be transferred (“performance obligations”), the Company can determine the transaction price for the goods or services to be transferred, the contract has commercial substance and it is probable that the Company will collect the consideration to which it will be entitled in exchange for the goods or services that will be transferred to the customer. Revenues are recorded in the amount of consideration to which the Company expects to be entitled in exchange for performance obligations upon transfer of control to the customer. 1. Revenues from selling products The Company recognizes revenues from selling goods at a point in time when control over the product is transferred to customers (upon delivery), at the net selling price, which reflects reserves for variable consideration, potential discounts and allowances. The transaction price is the consideration to which the Company expects to be entitled from the customer. The consideration promised in a contract with the Company’s customers may include fixed amounts and variable amounts. The Company estimates the variable consideration and includes it in the transaction price using the most likely outcome method, and only to the extent it is highly probable that a significant reversal of cumulative revenue recognized will not occur when the uncertainty associated with the variable consideration is subsequently resolved. Prior to recognizing revenue from variable consideration, the Company uses significant judgment to determine the probability of significant reversal of such revenue.
The Company has identified two performance obligations in the Chiesi Agreements as follows: (i) the license and research and development services and (ii) the contingent performance obligation regarding future manufacturing. The Company determined that the license together with the research and development services should be combined into single performance obligation since Chiesi cannot benefit from the license without the research and development services. The research and development services are highly specialized and are dependent on the supply of the drug. The manufacturing was contingent on regulatory approvals of the drug and the Company deems these services to be separately identifiable from other performance obligations in the contract. Manufacturing services post-regulatory approval are not interdependent or interrelated with the license, research and development services. Following the regulatory approvals for Elfabrio received in May 2023, the Company started recognizing revenue from manufacturing, see also revenue from selling products above. The transaction price was comprised of fixed consideration and variable consideration (capped research and development reimbursements). Under ASC 606, the consideration to which the Company would be entitled upon the achievement of contractual milestones, which are contingent upon the occurrence of future events, are a form of variable consideration. The Company estimates variable consideration using the most likely method. Amounts included in the transaction price are recognized only when it is probable that a significant reversal of cumulative revenues will not occur. Prior to recognizing revenue from variable consideration, the Company uses significant judgment to determine the probability of significant reversal of such revenue. Following the approval of Elfabrio by the FDA, the Company received a milestone payment equal to $20.0 million (see also note 4). Since the customer benefits from the research and development services as the entity performs the service, revenue from granting the license and the research and development services was recognized over time using the cost-to-cost method. Revenue from additional research and development services ordered by Chiesi is recognized over time using the cost-to-cost method.
Revenue from the research and development services was recognized over time using the cost-to-cost method since the customer benefits from the research and development services as the entity performs the services.
|
INVENTORIES |
9 Months Ended | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Sep. 30, 2023 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| INVENTORIES | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| INVENTORIES | NOTE 2 - INVENTORIES Inventories at September 30, 2023 and December 31, 2022 consisted of the following:
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||
FAIR VALUE MEASUREMENT |
9 Months Ended | |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Sep. 30, 2023 | ||||||||||||||||||||||||||||||||||||||||||||||||||
| FAIR VALUE MEASUREMENT | ||||||||||||||||||||||||||||||||||||||||||||||||||
| FAIR VALUE MEASUREMENT | NOTE 3 – FAIR VALUE MEASUREMENT The Company measures fair value and discloses fair value measurements for financial assets and liabilities. Fair value is based on the price that would be received from the sale of an asset, or paid to transfer a liability, in an orderly transaction between market participants at the measurement date. The accounting standard establishes a fair value hierarchy that prioritizes observable and unobservable inputs used to measure fair value into three broad levels, which are described below: Level 1: Quoted prices (unadjusted) in active markets that are accessible at the measurement date for assets or liabilities. The fair value hierarchy gives the highest priority to Level 1 inputs. Level 2: Observable prices that are based on inputs not quoted on active markets, but corroborated by market data. Level 3: Unobservable inputs are used when little or no market data is available. The fair value hierarchy gives the lowest priority to Level 3 inputs. In determining fair value, the Company utilizes valuation techniques that maximize the use of observable inputs and minimize the use of unobservable inputs to the extent possible and considers counterparty credit risk in its assessment of fair value. The fair value of the financial instruments included in the working capital of the Company is usually identical or close to their carrying value. Based on a Level 3 measurement, as of September 30, 2023, the fair value of the $20.4 million aggregate principal amount of the Company’s outstanding 2024 Notes is approximately $24.5 million. The value of these notes was estimated by implementing the binomial model. The liability component was valued based on the Income Approach. The following parameters were used:
|
REVENUES |
9 Months Ended | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Sep. 30, 2023 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| REVENUES | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| REVENUES | NOTE 4 – REVENUES The following table summarizes the Company’s disaggregation of revenues:
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
STOCK TRANSACTIONS |
9 Months Ended | |||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Sep. 30, 2023 | ||||||||||||||||||||||||||||||||||||||||||||
| STOCK TRANSACTIONS | ||||||||||||||||||||||||||||||||||||||||||||
| STOCK TRANSACTIONS | NOTE 5 – STOCK TRANSACTIONS
On June 28, 2023, the Company held its 2023 Annual Meeting of Stockholders, which was adjourned and reconvened on July 13, 2023 (the “Annual Meeting”). At the Annual Meeting, the Company’s stockholders, among other matters, approved an amendment to the Company’s Certificate of Incorporation, as amended, to increase the number of shares of Common Stock authorized for issuance from 144,000,000 to 185,000,000 (the “Charter Amendment”). The Charter Amendment was filed with the Secretary of State of the State of Delaware on July 25, 2023.
During the nine months ended September 30, 2023, the Company sold, in the aggregate, 12,560,150 shares of Common Stock under the 2023 Sales Agreement. The Company generated aggregate gross proceeds equal to approximately $24.9 million in connection with such sales. All such sales were completed during the quarters ended March 31, 2023 and June 30, 2023. No sales were completed during the three-months ended September 30, 2023. (c)Exercise of Warrants On May 8, 2023, the Company issued 301,810 shares of Common Stock in connection with the cash exercise of a warrant issued on March 18, 2020, as part of the Company’s private placement of Common Stock and warrants. The Company generated net proceeds equal to $0.7 million from the exercise of the warrant. On May 10, 2023, the Company issued 237,012 shares of Common Stock in connection with the cashless exercise of a warrant to purchase 845,000 shares of Common Stock issued on March 18, 2020, as part of the Company’s private placement of Common Stock and warrants. The Company did not generate any proceeds from the cashless exercise. No warrants were exercised during the three months ended September 30, 2023. (d)Conversion of 2024 Notes During the nine months ended September 30, 2023, the Company issued, in the aggregate, 4,691,623 shares of Common Stock in connection with the conversions of 2024 Notes. In connection with such conversions, during the nine months ended September 30, 2023, the Company paid to the converting holders $0.9 million representing cash payments due to accrued but unpaid interest, make-whole interest payments and payments in lieu of fractional shares. As a result of the conversions, the total principal amount of the 2024 Notes decreased by approximately $8.3 million. No 2024 Notes were converted during the three months ended September 30, 2023. (e)Stock based compensation
On September 14, 2023, the Company granted, with the approval of the Company’s compensation committee, 10-year options to purchase 85,715 shares of Common Stock, in the aggregate, to the new Chairman of the Company’s Board of Directors under the Plan. The options have an exercise price equal to $1.75 per share and vest over a three-year period in 12 equal quarterly increments. Vesting of the options is subject to automatic acceleration in full upon a Corporate Transaction or a Change in Control, as those terms are defined in the Plan, and are subject to certain other terms and conditions. The Company estimated the aggregate fair value of the options on the date of grant using the Black-Scholes option-pricing model to be approximately $0.1 million. On September 29, 2023, the Company granted, with the approval of the Company’s compensation committee, 10-year options to purchase 308,380 shares of Common Stock, in the aggregate, to the non-executive directors of the Company’s Board of Directors, other than the Chairman, under the Plan. The options have an exercise price equal to $1.66 per share and vest over a three-year period in 12 equal quarterly increments. Vesting of the options is subject to automatic acceleration in full upon a Corporate Transaction or a Change in Control, as those terms are defined in the Plan, and are subject to certain other terms and conditions. The Company estimated the aggregate fair value of the options on the date of grant using the Black-Scholes option-pricing model to be approximately $0.4 million. The fair value of each option granted during the three-month period ended September 30, 2023 is estimated at the date of grant using the Black-Scholes option-pricing model. The following weighted average assumptions were applied in determining the fair value of the options described in this Note 5(e)(3) on their respective grant dates:
|
EARNINGS (LOSS) PER SHARE |
9 Months Ended | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Sep. 30, 2023 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| EARNINGS (LOSS) PER SHARE | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| EARNINGS (LOSS) PER SHARE | NOTE 6 – EARNINGS (LOSS) PER SHARE Basic earnings (loss) per share is calculated by dividing the net income (loss) by the weighted average number of shares of Common Stock outstanding during each period. Diluted earnings per share is calculated by dividing the net income by the weighted-average number of shares of Common Stock outstanding during each period increased to include the number of additional shares of Common Stock that would have been outstanding if the potentially dilutive shares had been issued. In computing diluted earnings per share, basic earnings per share are adjusted to take into account the potential dilution that could occur upon: (i) the exercise of options granted under employee stock compensation plans using the treasury stock method; (ii) the exercise of warrants using the treasury stock method; and (iii) the conversion of the convertible notes using the “if-converted” method. Basic and diluted net earnings (loss) per share attributable to common stockholders were calculated as follows:
* Financial expenses on 2024 Notes consists of add back of financial expense incurred during the period and inclusion of make-whole interest payments that will be incurred upon conversion. Diluted earnings (loss) per share do not include 19,429,910 and 18,399,260 shares of Common Stock underlying outstanding stock options and warrants of the Company for the three and nine months ended September 30, 2023, respectively, because the effect would be anti-dilutive. Diluted earnings (loss) per share do not include 33,922,624 and 33,295,154 shares of Common Stock underlying outstanding stock options, warrants and the 2024 Notes for the three and nine months ended September 30, 2022, respectively, because the effect would be anti-dilutive. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TAXES ON INCOME |
9 Months Ended | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Sep. 30, 2023 | |||||||||||||||||||||||||||||||||||||||||||
| TAXES ON INCOME | |||||||||||||||||||||||||||||||||||||||||||
| TAXES ON INCOME | NOTE 7 – TAXES ON INCOME The following table summarizes the Company’s taxes on income:
The Company had an effective tax rate of 4% for the nine months ended September 30, 2023, compared to an effective tax rate of 0% for the nine months ended September 30, 2022. For the nine months ended September 30, 2023, the difference between the Company’s effective tax rate and the U.S. federal statutory rate of 21% was the result of the provision for current taxes on income mainly derived from U.S. taxable GILTI income mainly in respect of milestone payments and Section 174 of the U.S. Tax Cuts and Jobs Act, which was enacted in December 2017 (the “TCJA”), partially offset by the release of the valuation allowance on net operating losses (NOLs) in the United States. Following the regulatory approvals for Elfabrio in May 2023, the receipt of the $20.0 million milestone payment and the launch of Elfabrio in the United States, the Company released valuation allowance previously recorded on deferred tax assets in respect of its NOLs in the United States resulting in a net tax benefit of $3.1 million. The Company concluded that, based upon the preponderance of positive evidence over negative evidence and the anticipated ability to use the deferred tax assets, it was more likely than not that these deferred tax assets would be realizable due to forecasted profits. The Company considered the following: (i) cumulative profits for tax over the previous 12 quarters in its U.S. operations; (ii) the impact of Section 174 of the TCJA which requires the Company to capitalize and amortize its research and development expenses over 15 years; and (iii) its forecasted profits in the United States following the regulatory approvals of Elfabrio. |
||||||||||||||||||||||||||||||||||||||||||
SUPPLEMENTARY FINANCIAL STATEMENT INFORMATION |
9 Months Ended | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Sep. 30, 2023 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| SUPPLEMENTARY FINANCIAL STATEMENT INFORMATION | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| SUPPLEMENTARY FINANCIAL STATEMENT INFORMATION | NOTE 8 – SUPPLEMENTARY FINANCIAL STATEMENT INFORMATION Balance sheets:
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
SUBSEQUENT EVENTS |
9 Months Ended | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
Sep. 30, 2023 | ||||||||||
| SUBSEQUENT EVENTS | ||||||||||
| SUBSEQUENT EVENTS | NOTE 9 – SUBSEQUENT EVENTS
|
SIGNIFICANT ACCOUNTING POLICIES (Policies) |
9 Months Ended | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
Sep. 30, 2023 | ||||||||||
| SIGNIFICANT ACCOUNTING POLICIES | ||||||||||
| General |
Protalix BioTherapeutics, Inc. (collectively with its subsidiaries, the “Company”) and its wholly-owned subsidiaries, Protalix Ltd. and Protalix B.V. (collectively, the “Subsidiaries”), are biopharmaceutical companies focused on the development and commercialization of recombinant therapeutic proteins based on the Company’s proprietary ProCellEx® protein expression system (“ProCellEx”). The Company’s current focus is to facilitate the commercialization efforts of Chiesi Farmaceutici S.p.A. (“Chiesi”), the Company’s development and commercialization partner for pegunigalsidase alfa, or Elfabrio® (which the Company referred to as PRX-102 during its development stage), for the treatment of Fabry disease, a rare, genetic lysosomal disorder. To date, the Company has successfully developed taliglucerase alfa (marketed under the name Elelyso® except in Brazil where it is marketed as BioManguinhos alfataliglicerase), an enzyme replacement therapy for the treatment of Gaucher disease that has been approved for marketing in the United States, Brazil, Israel and other markets. The Company’s strategy is to develop proprietary recombinant proteins designed to address high, unmet needs in the rare disease space that are therapeutically superior to existing recombinant proteins currently marketed for the same indications. Consistent with this strategy, the Company has a number of product candidates in varying stages of the clinical development process. On May 5, 2023, the European Commission (“EC”) announced that it had approved the Marketing Authorization Application (“MAA”) for Elfabrio and on May 9, 2023, the U.S. Food and Drug Administration (“FDA”) announced that it had approved the Biologics License Application (“BLA”) for Elfabrio, each for adult patients with Fabry disease. Both approvals cover the 1 mg/kg every two weeks dosage. The European Medicines Agency (“EMA”) approval followed the February 2023 adoption of a positive opinion and recommendation of marketing authorization for Elfabrio by the EMA’s Committee for Medicinal Products for Human Use the (“CHMP”). Elfabrio was approved by the FDA with a boxed warning for hypersensitivity reactions/anaphylaxis, consistent with Enzyme Replacement Therapy (ERT) class labeling, and Warnings/Precautions providing guidance on the signs and symptoms of hypersensitivity and infusion-associated reactions seen in the clinical studies as well as treatments to manage such events should they occur. The Warnings/Precautions for membranoproliferative glomerulonephritis (MPGN) alert prescribers to the possibility of MPGN and provide guidance for appropriate patient management. Overall, the FDA review team concluded that in the context of Fabry disease as a rare, serious disease with limited therapeutic options that may not be suitable to all individual patients, the benefit-risk of Elfabrio is favorable for the treatment of adults with confirmed Fabry disease. In August and September of 2023, Elfabrio was approved in Great Britain and Switzerland, respectively, for long-term enzyme replacement therapy in adult patients with a confirmed diagnosis of Fabry disease. The Company has entered into two exclusive global licensing and supply agreements for Elfabrio with its development and commercialization partner for PRX-102, Chiesi Farmaceutici S.p.A. (“Chiesi”). On October 19, 2017, Protalix Ltd., the Company’s wholly-owned subsidiary, entered into an Exclusive License and Supply Agreement with Chiesi (the “Chiesi Ex-US Agreement”), pursuant to which Chiesi was granted an exclusive license for all markets outside of the United States to commercialize Elfabrio. On July 23, 2018, Protalix Ltd. entered into an Exclusive License and Supply Agreement with Chiesi (the Chiesi US Agreement, with respect to the commercialization of Elfabrio in the United States. Elfabrio, an enzyme replacement therapy, or ERT, was the subject of a phase III clinical program studying the drug as a treatment of patients with Fabry disease, a rare, genetic lysosomal disorder. The phase III clinical program included three separate studies, which are referred to as the BALANCE study, the BRIDGE study and the BRIGHT study. The phase III clinical program analyzed two potential dosing regimens: 1 mg/kg every two weeks and 2 mg/kg every four weeks. In addition, the phase III clinical program included two extension studies in which subjects that participated in our phase I/II clinical trials and phase III clinical trials had the opportunity to enroll and continue to be treated with PRX-102. As of March 1, 2023, sponsorship of the two open-label extension studies was transferred to Chiesi, which is now administering the extension studies. Over time, and as Elfabrio is approved for marketing in different jurisdictions, participants withdraw from the open-label extension studies. Some of the withdrawals transfer to a commercial setting, others withdraw for other reasons. The BLA for Elfabrio for the treatment of adult patients with Fabry disease was resubmitted to the FDA on November 9, 2022. An initial BLA for Elfabrio was submitted to the FDA on May 27, 2020 under the FDA’s Accelerated Approval pathway, but resulted in a Complete Response Letter (“CRL”). The MAA was submitted to the EMA on February 7, 2022, after the October 8, 2021 meeting we held, together with Chiesi, with the Rapporteur and Co-Rapporteur of the EMA regarding PRX-102. The FDA publicly released the internal review documents for Elfabrio (pegunigalsidase alfa-iwxj) injection BLA 761161. These documents provide previously unavailable additional information regarding the basis for the FDA’s May 2023 approval decision. In particular, the FDA determined that substantial evidence of effectiveness for Elfabrio in Fabry patients was established with one adequate and well-controlled study (Study PB-102-F01/02) with confirmatory evidence provided by the BALANCE study (also referred to as Study PB-102-F20). The FDA review team also concluded that the BALANCE study met its primary efficacy endpoint, which assessed the annualized rate of change in eGFR (estimated glomerular filtration rate) over 104 weeks. However, the FDA also determined that the results from the BALANCE study did not support a non-inferiority claim to the comparator product due to the lack of data to support a non-inferiority margin. The Company has licensed the rights to commercialize taliglucerase alfa worldwide (other than Brazil) to Pfizer Inc. (“Pfizer”), and in Brazil to Fundação Oswaldo Cruz (“Fiocruz”), an arm of the Brazilian Ministry of Health (the “Brazilian MoH”). Otherwise, except with respect to taliglucerase alfa and Elfabrio, the Company holds the worldwide commercialization rights to its other proprietary development candidates. In addition, the Company continuously evaluates potential strategic marketing partnerships as well as collaboration programs with biotechnology and pharmaceutical companies and academic research institutions. The Company’s product pipeline currently includes, among other candidates: (1)PRX-115, the Company’s plant cell-expressed recombinant PEGylated uricase (urate oxidase) – a chemically modified enzyme to treat severe gout; and (2)PRX-119, the Company’s plant cell-expressed PEGylated recombinant human DNase I product candidate being designed to elongate half-life in the circulation for NETs-related diseases. On March 21, 2023, the first patient was dosed in the Company’s phase I First-in-Human (“FIH”) clinical trial of PRX-115. As of September 30, 2023, 32 patients have been dosed in this trial. Obtaining marketing approval with respect to any product candidate in any country is dependent on the Company’s ability to implement the necessary regulatory steps required to obtain such approvals, and demonstrate the safety and efficacy of its product candidates. The Company cannot reasonably predict the outcome of these activities. On July 2, 2021, the Company entered into an At The Market Offering Agreement (the “2021 Sales Agreement”) with H.C. Wainwright & Co., LLC, as the Company’s sales agent (the “Agent”) which was amended on May 2, 2022. Pursuant to the terms of the 2021 Sales Agreement, the Company was able to sell, from time to time through the Agent, shares of its common stock, par value $0.001 per share (the “Common Stock”), having an aggregate offering price of up to $20.0 million (the “ATM Shares”). Upon execution of the 2021 Sales Agreement, the Company terminated the ATM Equity OfferingSM Sales Agreement it had entered into on October 1, 2020 with BofA Securities, Inc. (“BofA Securities”). During the term of the sales agreement with BofA Securities, the Company sold a total of 3,296,123 shares of Common Stock for total gross proceeds of approximately $13.8 million. During the term of the 2021 Sales Agreement which ended during the quarter ended March 31, 2023, the Company sold a total of 13,980,060 ATM Shares for total gross proceeds of approximately $20.0 million under the 2021 Sales Agreement, thereby completing the ATM program under said agreement. On February 27, 2023, the Company entered into an At The Market Offering Agreement (the “2023 Sales Agreement”) with the Agent. Pursuant to the terms of the 2023 Sales Agreement, the Company may sell, from time to time through the Agent, ATM Shares having an aggregate offering price of up to $20.0 million. As of September 30, 2023, shares of Common Stock for total gross proceeds of approximately $6.4 million remain available to be sold under the 2023 Sales Agreement. Under each of the Chiesi Ex-US Agreement and the Chiesi US Agreement (collectively, the “Chiesi Agreements”), Chiesi made an upfront payment to Protalix Ltd. of $25.0 million in connection with the execution of each agreement. In addition, under the Chiesi Ex-US Agreement, Protalix Ltd. is entitled to additional payments of up to $25.0 million in pegunigalsidase alfa development costs and to receive additional payments of up to $320.0 million, in the aggregate, in regulatory and commercial milestone payments. Under the Chiesi US Agreement, Protalix Ltd. is entitled to payments of up to a maximum of $20.0 million to cover development costs for pegunigalsidase alfa and to receive additional payments of up to a maximum of $760.0 million, in the aggregate, in regulatory and commercial milestone payments. To date, Protalix Ltd. has received the complete amount of development costs to which it is entitled under the Chiesi Agreements. In addition, following the approval of Elfabrio by the FDA, the Company received a milestone payment equal to $20.0 million. Under the terms of both of the Chiesi Agreements, Protalix Ltd. is required to manufacture all of the Elfabrio drug substance needed under the agreements, subject to certain exceptions, and Chiesi will purchase Elfabrio drug product from Protalix, subject to certain terms and conditions. The consideration for Protalix Ltd. is based on the drug product supplied to Chiesi and the average selling price of the drug product in the relevant territory multiplied by tiered payments as described in the relevant agreement. Under the Chiesi Ex-US Agreement, Chiesi is required to make tiered payments for drug product purchased from Protalix of 15% to 35%, depending on the amount of annual net sales outside of the United States, and under the Chiesi US Agreement, Chiesi is required to make tiered payments for drug product purchased from Protalix of 15% to 40%, depending on the amount of annual net sales in the United States. On August 29, 2022, the Company entered into a Fill/Finish Agreement (the “F/F Agreement”) and a Letter Agreement (the “Letter Agreement”), in each case with Chiesi. The Company agreed to supply Chiesi with drug substance for PRX-102 and, following relevant technology and technical information transfer activities, Chiesi has agreed, among other things, to provide the Company with commercial fill/finish services for PRX-102, including to support the anticipated global launch of PRX-102. The F/F Agreement shall continue in force until December 31, 2025, unless terminated earlier in accordance with the terms of the F/F Agreement and the term may be extended by mutual agreement for an additional period of seven years upon mutual written agreement prior to expiration of the initial term. On May 13, 2021, the Company signed a binding term sheet with Chiesi pursuant to which the Company and Chiesi amended the Chiesi Agreements in order to provide the Company with near-term capital. Chiesi agreed to make a $10.0 million payment to the Company before the end of the second quarter of 2021 in exchange for a $25.0 million reduction in a longer term regulatory milestone payment provided in the Chiesi EX-US Agreement. All other regulatory and commercial milestone payments remain unchanged. The Company received the payment in June 2021. The Company also agreed to negotiate certain manufacturing related matters. Since its approval by the FDA, taliglucerase alfa has been marketed by Pfizer in accordance with the exclusive license and supply agreement entered into between Protalix Ltd. and Pfizer, which is referred to herein as the Pfizer Agreement. In October 2015, Protalix Ltd. and Pfizer entered into an amended exclusive license and supply agreement, which is referred to herein as the Amended Pfizer Agreement, pursuant to which the Company sold to Pfizer its share in the collaboration created under the Pfizer Agreement for the commercialization of Elelyso. As part of the sale, the Company agreed to transfer its rights to Elelyso in Israel to Pfizer while gaining full rights to it in Brazil. Under the Amended Pfizer Agreement, Pfizer is entitled to all of the revenues, and is responsible for 100% of expenses globally for Elelyso, excluding Brazil where the Company is responsible for all expenses and retains all revenues. On June 18, 2013, the Company entered into a Supply and Technology Transfer Agreement with Fiocruz (the “Brazil Agreement”) for taliglucerase alfa. Fiocruz’s purchases of BioManguinhos alfataliglicerase to date have been significantly below certain agreed-upon purchase milestones and, accordingly, the Company has the right to terminate the Brazil Agreement. Notwithstanding the termination right, the Company is, at this time, continuing to supply BioManguinhos alfataliglicerase to Fiocruz and patients continue to be treated with BioManguinhos alfataliglicerase in Brazil. The Company expects to continue to incur significant expenditures in the near future due to research and developments efforts with respect to the product candidates. Under the terms of the Company’s outstanding 7.50% Senior Secured Convertible Notes due 2024 (the “2024 Notes”), the Company is required to comply with certain financial covenants, including the maintenance of a minimum cash balance of at least $7.5 million. As of September 30, 2023, the Company is in compliance with all such covenants. The Company believes that its cash and cash equivalents as of September 30, 2023, are sufficient to satisfy the Company’s capital needs for at least 12 months from the date that these financial statements are issued. |
|||||||||
| Basis of presentation |
The accompanying unaudited condensed consolidated financial statements of the Company have been prepared in accordance with generally accepted accounting principles in the United States (“GAAP”) for interim financial information. Accordingly, they do not include all of the information and notes required by GAAP for annual financial statements. In the opinion of management, all adjustments (of a normal recurring nature) considered necessary for a fair statement of the results for the interim periods presented have been included. Operating results for the interim period are not necessarily indicative of the results that may be expected for the full year. These unaudited condensed consolidated financial statements should be read in conjunction with the audited consolidated financial statements in the Annual Report on Form 10-K for the year ended December 31, 2022, filed by the Company with the U.S. Securities and Exchange Commission (the “Commission”). The comparative balance sheet at December 31, 2022 has been derived from the audited financial statements at that date. There have been no material changes in our significant accounting policies as described in our consolidated financial statements for the year ended December 31, 2022. |
|||||||||
| Revenue recognition |
The Company accounts for revenue pursuant to Accounting Standards Codification, Topic 606, Revenue from Contracts with Customers (“ASC 606”). Under ASC 606, a contract with a customer exists only when: the parties to the contract have approved it and are committed to perform their respective obligations, the Company can identify each party’s rights regarding the distinct goods or services to be transferred (“performance obligations”), the Company can determine the transaction price for the goods or services to be transferred, the contract has commercial substance and it is probable that the Company will collect the consideration to which it will be entitled in exchange for the goods or services that will be transferred to the customer. Revenues are recorded in the amount of consideration to which the Company expects to be entitled in exchange for performance obligations upon transfer of control to the customer. 1. Revenues from selling products The Company recognizes revenues from selling goods at a point in time when control over the product is transferred to customers (upon delivery), at the net selling price, which reflects reserves for variable consideration, potential discounts and allowances. The transaction price is the consideration to which the Company expects to be entitled from the customer. The consideration promised in a contract with the Company’s customers may include fixed amounts and variable amounts. The Company estimates the variable consideration and includes it in the transaction price using the most likely outcome method, and only to the extent it is highly probable that a significant reversal of cumulative revenue recognized will not occur when the uncertainty associated with the variable consideration is subsequently resolved. Prior to recognizing revenue from variable consideration, the Company uses significant judgment to determine the probability of significant reversal of such revenue.
The Company has identified two performance obligations in the Chiesi Agreements as follows: (i) the license and research and development services and (ii) the contingent performance obligation regarding future manufacturing. The Company determined that the license together with the research and development services should be combined into single performance obligation since Chiesi cannot benefit from the license without the research and development services. The research and development services are highly specialized and are dependent on the supply of the drug. The manufacturing was contingent on regulatory approvals of the drug and the Company deems these services to be separately identifiable from other performance obligations in the contract. Manufacturing services post-regulatory approval are not interdependent or interrelated with the license, research and development services. Following the regulatory approvals for Elfabrio received in May 2023, the Company started recognizing revenue from manufacturing, see also revenue from selling products above. The transaction price was comprised of fixed consideration and variable consideration (capped research and development reimbursements). Under ASC 606, the consideration to which the Company would be entitled upon the achievement of contractual milestones, which are contingent upon the occurrence of future events, are a form of variable consideration. The Company estimates variable consideration using the most likely method. Amounts included in the transaction price are recognized only when it is probable that a significant reversal of cumulative revenues will not occur. Prior to recognizing revenue from variable consideration, the Company uses significant judgment to determine the probability of significant reversal of such revenue. Following the approval of Elfabrio by the FDA, the Company received a milestone payment equal to $20.0 million (see also note 4). Since the customer benefits from the research and development services as the entity performs the service, revenue from granting the license and the research and development services was recognized over time using the cost-to-cost method. Revenue from additional research and development services ordered by Chiesi is recognized over time using the cost-to-cost method.
Revenue from the research and development services was recognized over time using the cost-to-cost method since the customer benefits from the research and development services as the entity performs the services. |
INVENTORIES (Tables) |
9 Months Ended | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Sep. 30, 2023 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| INVENTORIES | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Schedule of Inventory |
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||
FAIR VALUE MEASUREMENT (Tables) |
9 Months Ended | |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Sep. 30, 2023 | ||||||||||||||||||||||||||||||||||||||||||||||||||
| 2024 Notes | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Schedule of Fair Value Assumptions |
|
REVENUES (Tables) |
9 Months Ended | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Sep. 30, 2023 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| REVENUES | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Schedule of Company's Disaggregation of Revenues |
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
STOCK TRANSACTIONS (Tables) |
9 Months Ended | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Sep. 30, 2023 | |||||||||||||||||||||||||||||
| STOCK TRANSACTIONS | |||||||||||||||||||||||||||||
| Schedule of Stock Option Valuation Assumptions |
|
EARNINGS (LOSS) PER SHARE (Tables) |
9 Months Ended | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Sep. 30, 2023 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| EARNINGS (LOSS) PER SHARE | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Schedule of Basic and Diluted Net Earnings (Loss) Per Share |
* Financial expenses on 2024 Notes consists of add back of financial expense incurred during the period and inclusion of make-whole interest payments that will be incurred upon conversion. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TAXES ON INCOME (Tables) |
9 Months Ended | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Sep. 30, 2023 | |||||||||||||||||||||||||||||||||||||||||||
| TAXES ON INCOME | |||||||||||||||||||||||||||||||||||||||||||
| Schedule of components of Income Tax Expense (Benefit) |
|
||||||||||||||||||||||||||||||||||||||||||
SUPPLEMENTARY FINANCIAL STATEMENT INFORMATION (Tables) |
9 Months Ended | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Sep. 30, 2023 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| SUPPLEMENTARY FINANCIAL STATEMENT INFORMATION | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Supplemental Information, Balance Sheets |
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
SIGNIFICANT ACCOUNTING POLICIES (Details) $ / shares in Units, $ in Thousands |
1 Months Ended | 3 Months Ended | 9 Months Ended | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Feb. 27, 2023
USD ($)
|
Aug. 29, 2022 |
Jul. 02, 2021
USD ($)
$ / shares
shares
|
May 13, 2021
USD ($)
|
Jul. 23, 2018
USD ($)
|
Oct. 19, 2017
USD ($)
|
Oct. 31, 2015 |
Sep. 30, 2023
USD ($)
agreement
$ / shares
shares
|
Mar. 31, 2023
USD ($)
shares
|
Sep. 30, 2023
USD ($)
agreement
$ / shares
shares
|
Sep. 30, 2022
USD ($)
|
Mar. 21, 2023
item
|
Dec. 31, 2022
$ / shares
|
|
| Significant Accounting Policies [Line Items] | |||||||||||||
| Number of global licensing and supply agreements | agreement | 2 | 2 | |||||||||||
| Number of patients dosed | item | 32 | ||||||||||||
| Common Stock, Par or Stated Value Per Share | $ / shares | $ 0.001 | $ 0.001 | $ 0.001 | ||||||||||
| Stock issuance proceeds | $ 23,954 | $ 4,161 | |||||||||||
| Convertible Debt, Current | $ 20,192 | 20,192 | |||||||||||
| ATM Shares | |||||||||||||
| Significant Accounting Policies [Line Items] | |||||||||||||
| Number of shares issued (in shares) | shares | 13,980,060 | ||||||||||||
| Common Stock, Par or Stated Value Per Share | $ / shares | $ 0.001 | ||||||||||||
| Sale of stock, maximum offering price | $ 20,000 | $ 20,000 | $ 6,400 | ||||||||||
| Stock issuance proceeds | $ 20,000 | ||||||||||||
| ATM Equity Offering Sales Agreement | |||||||||||||
| Significant Accounting Policies [Line Items] | |||||||||||||
| Number of shares issued (in shares) | shares | 3,296,123 | 0 | 12,560,150 | ||||||||||
| Stock issuance proceeds | $ 13,800 | $ 24,900 | |||||||||||
| 2024 Notes | |||||||||||||
| Significant Accounting Policies [Line Items] | |||||||||||||
| Interest rate (as a percent) | 7.50% | 7.50% | |||||||||||
| Maintain of Minimum Cash Balance | $ 7,500 | $ 7,500 | |||||||||||
| Amended Pfizer Agreement | Protalix Bio Therapeutics Incorporation | Brazil | |||||||||||||
| Significant Accounting Policies [Line Items] | |||||||||||||
| Collaborative Arrangement Revenues and Expenses Sharing Percentage | 100.00% | ||||||||||||
| Chiesi Agreements | |||||||||||||
| Significant Accounting Policies [Line Items] | |||||||||||||
| Amount of milestone payment received | $ 20,000 | ||||||||||||
| Revenue, Performance Obligation, Number | agreement | 2 | 2 | |||||||||||
| Chiesi US Agreement | |||||||||||||
| Significant Accounting Policies [Line Items] | |||||||||||||
| Additional Amounts Payable To Cover Development Costs | $ 20,000 | ||||||||||||
| Additional Amount Payable For Achievement Of Regulatory And Commercial Milestones | $ 760,000 | ||||||||||||
| Chiesi US Agreement | Minimum | |||||||||||||
| Significant Accounting Policies [Line Items] | |||||||||||||
| Payment On Net Sales Percentage | 15.00% | ||||||||||||
| Chiesi US Agreement | Maximum | |||||||||||||
| Significant Accounting Policies [Line Items] | |||||||||||||
| Payment On Net Sales Percentage | 40.00% | ||||||||||||
| Chiesi Ex-US Agreement | |||||||||||||
| Significant Accounting Policies [Line Items] | |||||||||||||
| Additional Amounts Payable To Cover Development Costs | $ 25,000 | ||||||||||||
| Agreement Amendment Payment Receivable | $ 10,000 | ||||||||||||
| Change In Amount Receivable For Achievement Of Regulatory And Commercial Milestones | $ 25,000 | ||||||||||||
| Additional Amount Payable For Achievement Of Regulatory And Commercial Milestones | $ 320,000 | ||||||||||||
| Chiesi Ex-US Agreement | Minimum | |||||||||||||
| Significant Accounting Policies [Line Items] | |||||||||||||
| Payment On Net Sales Percentage | 15.00% | ||||||||||||
| Chiesi Ex-US Agreement | Maximum | |||||||||||||
| Significant Accounting Policies [Line Items] | |||||||||||||
| Payment On Net Sales Percentage | 35.00% | ||||||||||||
| Fill/Finish Agreement | |||||||||||||
| Significant Accounting Policies [Line Items] | |||||||||||||
| Extended term of agreement | 7 years | ||||||||||||
| Chiesi US Agreement and Chiesi Ex-US Agreement | |||||||||||||
| Significant Accounting Policies [Line Items] | |||||||||||||
| Non-refundable Payment Receivable | $ 25,000 | $ 25,000 | |||||||||||
INVENTORIES (Schedule of Inventory) (Details) - USD ($) $ in Thousands |
Sep. 30, 2023 |
Dec. 31, 2022 |
|---|---|---|
| INVENTORIES | ||
| Raw materials | $ 4,198 | $ 3,508 |
| Work in progress | 9,116 | 2,678 |
| Finished goods (drug substance and drug product) | 8,269 | 10,618 |
| Total inventory | $ 21,583 | $ 16,804 |
FAIR VALUE MEASUREMENT (Schedule of Fair Value Assumptions) (Details) - 2024 Notes - Level 3 |
Sep. 30, 2023
$ / shares
Y
|
|---|---|
| Share Price. | |
| Debt Instrument, Measurement Input | $ / shares | 1.66 |
| Expected term | |
| Debt Instrument, Measurement Input | Y | 0.93 |
| Risk free rate | |
| Debt Instrument, Measurement Input | 5.34 |
| Volatility | |
| Debt Instrument, Measurement Input | 62.84 |
| Yield | |
| Debt Instrument, Measurement Input | 12.49 |
FAIR VALUE MEASUREMENT (Additional Information) (Details) - Level 3 - 2024 Notes $ in Thousands |
Sep. 30, 2023
USD ($)
|
|---|---|
| Debt Instrument Carrying Amount | $ 20,400 |
| Debt Instrument Fair Value | $ 24,500 |
REVENUES (Details) - USD ($) $ in Thousands |
3 Months Ended | 9 Months Ended | ||
|---|---|---|---|---|
Sep. 30, 2023 |
Sep. 30, 2022 |
Sep. 30, 2023 |
Sep. 30, 2022 |
|
| Revenues | $ 10,345 | $ 14,183 | $ 55,008 | $ 39,021 |
| Goods | ||||
| Revenues | 10,168 | 8,812 | 30,309 | 21,222 |
| Goods | Pfizer | ||||
| Revenues | 2,304 | 4,541 | 7,972 | 11,279 |
| Goods | Brazil | ||||
| Revenues | 2,320 | 1,708 | 5,120 | 7,162 |
| Goods | Chiesi | ||||
| Revenues | 5,544 | 2,563 | 17,217 | 2,781 |
| License and R&D Services | ||||
| Revenues | $ 177 | $ 5,371 | $ 24,699 | $ 17,799 |
STOCK TRANSACTIONS (Details) - USD ($) $ in Thousands |
3 Months Ended | 9 Months Ended | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
May 10, 2023 |
May 08, 2023 |
Jul. 02, 2021 |
Sep. 30, 2023 |
Sep. 30, 2023 |
Sep. 30, 2022 |
Jul. 25, 2023 |
Jul. 24, 2023 |
Jun. 28, 2023 |
Jun. 27, 2023 |
Dec. 31, 2022 |
|
| Class of Stock [Line Items] | |||||||||||
| Common Stock, Shares Authorized | 185,000,000 | 185,000,000 | 185,000,000 | 144,000,000 | 144,000,000 | ||||||
| Exercise of warrants (in shares) | 237,012 | 0 | |||||||||
| Exercise of warrants | $ 712 | $ 2 | |||||||||
| Exercise of warrants | $ 700 | 712 | 2 | ||||||||
| Warrants issued to purchase common stock | 845,000 | ||||||||||
| Stock issuance proceeds | $ 23,954 | $ 4,161 | |||||||||
| 2006 Employee Stock Incentive Plan | |||||||||||
| Class of Stock [Line Items] | |||||||||||
| Number of shares authorized for issuance under share-based payment arrangement | 12,475,171 | 8,475,171 | |||||||||
| 2024 Notes | |||||||||||
| Class of Stock [Line Items] | |||||||||||
| Converted instrument, shares issued | 0 | 4,691,623 | |||||||||
| Paid to the converting holders | $ 900 | ||||||||||
| Decrease in principal amount | $ 8,300 | ||||||||||
| ATM Equity Offering Sales Agreement | |||||||||||
| Class of Stock [Line Items] | |||||||||||
| Number of shares issued (in shares) | 3,296,123 | 0 | 12,560,150 | ||||||||
| Stock issuance proceeds | $ 13,800 | $ 24,900 | |||||||||
| Private Placement | |||||||||||
| Class of Stock [Line Items] | |||||||||||
| Exercise of warrants (in shares) | 301,810 | ||||||||||
STOCK TRANSACTIONS - Stock based compensation (Details) $ / shares in Units, $ in Millions |
3 Months Ended | |||||
|---|---|---|---|---|---|---|
|
Sep. 29, 2023
USD ($)
item
$ / shares
shares
|
Sep. 14, 2023
USD ($)
item
$ / shares
shares
|
Aug. 15, 2023
USD ($)
item
$ / shares
shares
|
Sep. 30, 2023
$ / shares
|
Jun. 28, 2023
shares
|
Jun. 27, 2023
shares
|
|
| Options | ||||||
| Class of Stock [Line Items] | ||||||
| Exercise price | $ / shares | $ 1.34 | |||||
| 2006 Employee Stock Incentive Plan | ||||||
| Class of Stock [Line Items] | ||||||
| Number of shares authorized for issuance under share-based payment arrangement | 12,475,171 | 8,475,171 | ||||
| 2006 Employee Stock Incentive Plan | Shares of restricted Common Stock | ||||||
| Class of Stock [Line Items] | ||||||
| Number of shares granted | 1,371,362 | |||||
| Estimated fair value on date of grant | $ | $ 2.7 | |||||
| 2006 Employee Stock Incentive Plan | Shares of restricted Common Stock | Upon grant | ||||||
| Class of Stock [Line Items] | ||||||
| Number of shares granted | 200,000 | |||||
| 2006 Employee Stock Incentive Plan | Shares of restricted Common Stock | Over a two-year period | ||||||
| Class of Stock [Line Items] | ||||||
| Number of shares granted | 600,000 | |||||
| Vesting period | 2 years | |||||
| Vesting in number of quarterly increments | item | 8 | |||||
| 2006 Employee Stock Incentive Plan | Shares of restricted Common Stock | Over a three-year period | ||||||
| Class of Stock [Line Items] | ||||||
| Number of shares granted | 571,362 | |||||
| Vesting period | 3 years | |||||
| Vesting in number of quarterly increments | item | 12 | |||||
| 2006 Employee Stock Incentive Plan | Options | Officers and other employees | ||||||
| Class of Stock [Line Items] | ||||||
| Vesting period | 3 years | |||||
| Vesting in number of quarterly increments | item | 12 | |||||
| Estimated fair value on date of grant | $ | $ 1.5 | |||||
| Term of award | 10 years | |||||
| Number of options to purchase shares | 1,056,315 | |||||
| Exercise price | $ / shares | $ 1.99 | |||||
| 2006 Employee Stock Incentive Plan | Options | Chairman, Board of Directors | ||||||
| Class of Stock [Line Items] | ||||||
| Vesting period | 3 years | |||||
| Vesting in number of quarterly increments | item | 12 | |||||
| Estimated fair value on date of grant | $ | $ 0.1 | |||||
| Term of award | 10 years | |||||
| Number of options to purchase shares | 85,715 | |||||
| Exercise price | $ / shares | $ 1.75 | |||||
| 2006 Employee Stock Incentive Plan | Options | Non-executive directors other than Chairman | ||||||
| Class of Stock [Line Items] | ||||||
| Vesting period | 3 years | |||||
| Vesting in number of quarterly increments | item | 12 | |||||
| Estimated fair value on date of grant | $ | $ 0.4 | |||||
| Term of award | 10 years | |||||
| Number of options to purchase shares | 308,380 | |||||
| Exercise price | $ / shares | $ 1.66 |
STOCK TRANSACTIONS - Schedule of Weighted average assumptions (Details) - Options |
3 Months Ended |
|---|---|
|
Sep. 30, 2023
$ / shares
| |
| Class of Stock [Line Items] | |
| Weighted average grants date fair value (USD) | $ 1.34 |
| Exercise price (USD) | $ 1.91 |
| Risk free rate | 4.40% |
| Volatility | 79.40% |
| Dividend yield | 0.00% |
| Expected life (Years) | 5 years 9 months |
EARNINGS (LOSS) PER SHARE (Details) - USD ($) $ in Thousands |
3 Months Ended | 9 Months Ended | ||
|---|---|---|---|---|
Sep. 30, 2023 |
Sep. 30, 2022 |
Sep. 30, 2023 |
Sep. 30, 2022 |
|
| Numerator: | ||||
| Net income (loss) | $ (1,852) | $ (3,567) | $ 14,356 | $ (11,187) |
| Financial expenses of 2024 Notes | (1,766) | (1,610) | ||
| Net income (loss) for diluted calculation | $ (3,618) | $ (3,567) | $ 12,746 | $ (11,187) |
| Denominator: | ||||
| Weighted average shares of Common Stock outstanding for basic calculation | 72,281,681 | 49,498,105 | 65,811,506 | 47,582,733 |
| Weighted average dilutive effect of 2024 Notes | 11,500,998 | 13,981,660 | ||
| Weighted average dilutive effect of stock options | 1,247,115 | |||
| Weighted average shares of Common Stock outstanding for diluted calculation | 83,782,679 | 49,498,105 | 81,040,281 | 47,582,733 |
| Outstanding Stock Options And Warrants | ||||
| Denominator: | ||||
| Antidilutive securities excluded from computation of earnings per share, amount | 19,429,910 | 18,399,260 | ||
| Outstanding Stock Options Warrants And 2024 Notes | ||||
| Denominator: | ||||
| Antidilutive securities excluded from computation of earnings per share, amount | 33,922,624 | 33,295,154 | ||
TAXES ON INCOME (Details) - USD ($) $ in Thousands |
3 Months Ended | 9 Months Ended | ||
|---|---|---|---|---|
Sep. 30, 2023 |
Sep. 30, 2023 |
Sep. 30, 2022 |
May 31, 2023 |
|
| TAXES ON INCOME | ||||
| Current taxes on income | $ 95 | $ 3,728 | ||
| Deferred taxes on income | 38 | (3,092) | ||
| Total taxes on income | 133 | $ 636 | ||
| Effective income tax rate (as a percent) | 4.00% | 0.00% | ||
| Statutory income tax rate | 21.00% | |||
| Milestone Payments to regulatory approvals | $ 20,000 | |||
| Valuation allowance related to deferred tax assets | $ 3,100 | |||
SUPPLEMENTARY FINANCIAL STATEMENT INFORMATION (Accounts Payable and Accruals - Other) (Details) - USD ($) $ in Thousands |
Sep. 30, 2023 |
Dec. 31, 2022 |
|---|---|---|
| Accounts payable and accruals - other: | ||
| Payroll and related expenses | $ 1,239 | $ 1,216 |
| Interest Payable | 123 | 719 |
| Provision for vacation | 1,471 | 1,404 |
| Accrued expenses | 9,249 | 7,478 |
| Royalties payable | 332 | 781 |
| Income tax payable | 3,257 | 530 |
| Reserve for deductions from revenue | 2,816 | |
| Property and equipment suppliers | 253 | 143 |
| Accounts payable and accruals - other | $ 18,740 | $ 12,271 |
SUBSEQUENT EVENTS (Details) $ / shares in Units, $ in Thousands |
3 Months Ended | ||
|---|---|---|---|
|
Oct. 15, 2023
USD ($)
item
$ / shares
shares
|
Oct. 04, 2023
USD ($)
|
Sep. 30, 2023
$ / shares
|
|
| Stock Options | |||
| Subsequent Event [Line Items] | |||
| Exercise price | $ / shares | $ 1.34 | ||
| Subsequent Event | Chiesi Agreements | |||
| Subsequent Event [Line Items] | |||
| Proceeds From Sale Of Products | $ | $ 6,400 | ||
| Subsequent Event | Stock Options | New officer | 2006 Employee Stock Incentive Plan | |||
| Subsequent Event [Line Items] | |||
| Term of award | 10 years | ||
| Number of options to purchase shares | shares | 250,000 | ||
| Exercise price | $ / shares | $ 1.48 | ||
| Vesting period | 3 years | ||
| Vesting in number of quarterly increments | item | 12 | ||
| Estimated fair value on date of grant | $ | $ 260 |
1 Year Protalix BioTherapeutics Chart |
1 Month Protalix BioTherapeutics Chart |

It looks like you are not logged in. Click the button below to log in and keep track of your recent history.
Support: +44 (0) 203 8794 460 | support@advfn.com
By accessing the services available at ADVFN you are agreeing to be bound by ADVFN's Terms & Conditions
 Hot Features
Hot Features